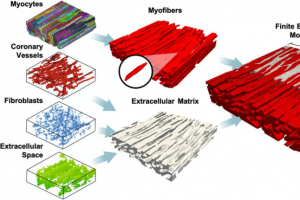
概述 肺动脉高压(PAH)等心脏疾病会造成心脏组织结构和力学行为发生实质性改变。PAH 使心脏右心室(RV)长期压力超负荷,右心室最初通过肌纤维肥大增厚以减轻壁应力的增加,但随后扩张并失去收缩功能,导致右心室衰竭。有研究表明,压力超负荷导致的右心室重构是右心室功能不良预后的主要预测因素之一。因此,量化右心室的力学状态,通过开发计算模型更好地了解 PAH 引起重塑的发生、发展和潜在可逆性的影响因素十分必要。 通过心脏的多尺度计算建模,将细胞尺度与组织尺度行为联系起来,可以提高对心脏重塑的理解,并更好地确定治疗靶点。本研究结合共聚焦显微镜技术、软组织力学和有限元建模,开发了一种高保真的心室心肌微观解剖学仿真模型。 亮点 基于兔心肌的高分辨率 3D 成像数据创建三维模型在Simpleware FE 中生成高质量的网格模型在 FEniCS 软件中进行有限元分析 图像处理 取直径为 5mm 的新西兰大白兔左心室心肌样本,冷冻 100 μm 厚的切片,标记切片并在 Fluoromount-G 内密封使其不受压。通过激光扫描共聚焦显微镜获得三维图像堆栈,成像组织体积为 204 × 204 × 60 μm。 采用分水岭算法和基于直方图的阈值分割半自动方法对三维组织结构进行分割和重建。为简化初始有限元模型的开发,将肌细胞连接并组合成“肌纤维”相,而不是单个肌细胞。冠状血管、成纤维细胞和细胞外空间被合并成“细胞外基质(ECM)”或“胶原”相,消除模型域中的空洞。在 Simpleware ScanIP 中利用 Island removal 去除孤岛,使用 Recursive Gaussian 滤波器平滑。在 Simpleware FE 模块中生成由约 110 万个线性四面体单元组成的体积网格。经过图像处理后,肌细胞的方向与 e1 轴对齐,交叉纤维方向与 e2 轴对齐。 图1:左图依次为心肌细胞、冠状动脉血管、成纤维细胞、细胞外空间。中图:肌细胞组成肌纤维相(红色),放大突出显示的为代表性肌细胞;冠状动脉血管、成纤维细胞和细胞外空间合并为细胞外基质相(灰色)。右图:FE 模型横截面展示肌纤维单元嵌入在 ECM单元中。 模拟 单层仿真 基于结构的模型最初是为了具有更分散纤维分布的组织尺度肌纤维/胶原蛋白/相互作用应力而开发。因此,作为微观解剖模型的拟合目标,使用微观解剖有限元几何高度对齐的结构和基于结构的模型模拟应力应变响应。在开源软件 FEniCS 中不同双轴应变配置 E11:E22 = 0.30:0.30、0.30:0.15 和 0.15:0.30 下,对模型的边界表面施加变形。 […]