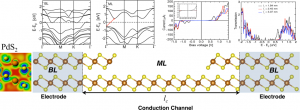
参考文献: Ghorbani-Asl, A. Kuc, P. Miró, and T. Heine, A Single-Material Logical Junction Based on 2D Crystal PdS2, Adv. Mater. Early View (2016) Thomas Heine和他的同事通过计算设计了一种由PdS2单独一种材料构成的逻辑结器件。非常独特的是,二维的过渡金属过硫化物PdS2 在单层的状态下具有半导体特性,在双层的状态下具有半金属特性。利用电子的这种相变,可以从双层PdS2制备出单层PdS2结器件。 在这项研究中,能带结构的计算使用了BAND模块中的自旋轨道耦合,相关输运计算使用了DFTB与非平衡格林函数方法。其中一个PdS2 结的表现出二极管的特征,有一个2.5纳米长的通道。 二维材料的研究刚刚兴起,二维电子学中的新型器件概念,还有待发掘。不同方式的结合可能会产生史无前例意想不到的纳米电子学性质。