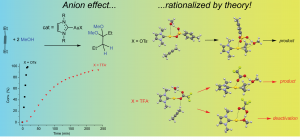
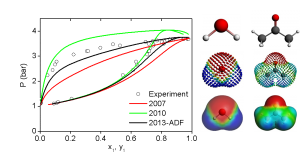
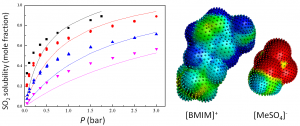
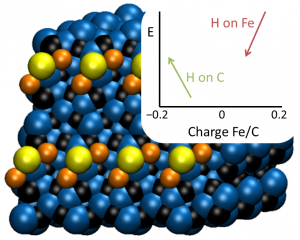
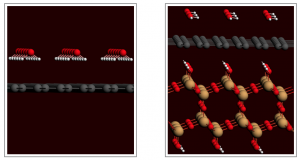
参考文献: G. Ciancaleoni, L. Belpassi, D. Zuccaccia, F. Tarantelli, and P. Belanzoni,Counterion Effect in the Reaction Mechanism of NHC Gold(I)-Catalyzed Alkoxylation of Alkynes: Computational Insight into Experiment, ACS Catal. 5, 803–814 (2015) 金催化在有机合成中,已经成了孤立π-体系亲核反应、C-H活化、交叉偶联反应、光催化以及不对称转化中非常流行的基本工具。和配体的作用类似,金催化平衡离子被认为是调制催化效果的基本要素。但催化循环反应的不同阶段,阴离子所起的作用还在争论之中。 从实验数据来看,阴离子倾向于作为配体和氢键受体,意想不到地增强L–Au–X (L = ligand, X = anion)催化炔的烷氧基化的效率,其中亲核进攻时决速步。 DFT计算显示,在这一步中,阴离子的作用是: 作为模板支撑外部环境亲核分子在适当位置进攻; 作为氢键受体,增强进攻的乙醇分子的亲核性; 通过强配位能力和/或碱度,钝化催化剂,阻止炔配位,或形成自由醇盐。 在质子去金化这一步,阴离子成了质子的“摆渡车”,从而降低了活化能垒。基于这种反应机理,DFT计算倾向于支持中间配位,以及碱性阴离子是最有效的催化剂。 甲苯磺酸盐平衡离子打破了它作为Au-NHC化合物配体以及氢键受体的完美平衡,增强了催化炔的烷氧基化的效果。 关键词:Relativistic DFT, reaction mechanisms, homogeneous catalysis