概述 ADF作为世界上最早的密度泛函软件,从上世纪70年代初诞生以来,在逐渐强大的开发团队的支持下,在计算理论、方法、计算分析工具、图形化用户界面方面,不断快速的发展。当初它只是一个非周期性体系(例如分子、团簇)的密度泛函计算程序。70年代,它的名字叫做HFS,后来更名为Amol,再后来更名为ADF。 90年代,创始团队(E.J. Baerends等)首次提出了相当精确的处理电子相对论效应(尤其是自旋轨道耦合)的理论 Zero Order Regular Approximation(ZORA),并将其在ADF中实现,成为至今最流行(没有之一)的处理自旋轨道耦合的方法。2017年,最新、最先进的相对论密度泛函方法X2C在ADF中实现。在计算化学领域中,精度与效率常常是一对矛盾,一般而言精度高则效率低,反之亦然。而X2C在精度与效率方面,皆远优于ZORA,是一个极为天才的全新方法。2000以后,更多的计算模块,例如自行开发的周期性体系密度泛函计算的BAND、流体热力学程序COSMO-RS,以及其他研究组发展的流行程序,如反应力场ReaxFF、Quantum Espresso、MOPAC、DFTB也逐渐纳入其中。功能模块越来越丰富,但此时整个软件平台的名字还是叫ADF,其中最早的模块也叫ADF。如同最开始一样,ADF并没有将自己定位为一个“程序打包商”。在计算理论与方法上,它从来没有停止过创新。能量分解方法(EDA)、成键分析工具ETS-NOCV等化学分析方法,不仅被集成到ADF模块中,还被创造性地应用于周期性体系的密度泛函计算中,使得BAND成为一个极具特色、功能强大的周期性体系化学问题研究工具。近两年BAND的功能突飞猛进,使用BAND完成的创造性的杰出工作开始出现在JACS、Angew. Chem. Int. Ed.等重要化学期刊。 为避免ADF软件平台与其中ADF模块重名,同时也因为其他模块越来越强大、完善,2018新版发布,ADF软件平台更名为Amsterdam Modeling Suite,简称AMS。各个模块名字,如ADF、BAND、COSMO-RS等保持不变。 AMS 2018 新功能概览: Amsterdam Density Functional (ADF) 在考虑COSMO溶剂化模型,或者使用某些range-separated泛函时,也可以进行激发态结构优化、频率、Linear transit、过渡态搜索等计算 激发态分析:电荷转移描述符、片段分析、eXcited Constrained DFT、电荷转移 其他分析工具:移除空轨道的能量分解、NBO6更新升级 新的快速计算金、银团簇体系紫外可见吸收光谱的方法:POLTDDFT Ab Initio分子动力学 Periodic DFT: BAND 与 Quantum ESPRESSO 弹性张量及其相关性质,例如体积模量 分子动力学 Linear transit与势能面扫描(PES) Crystal Orbital Overlap Populations (COOP) LDOS(STM成像) 外加压力下的结构优化 任意k点有效质量 自洽迭代改善 Quantum ESPRESSO版本更新到6.3版,包含CPMD功能 ReaxFF 新的加速反应的方法:Collective Variable […]


材料与器件模拟研讨会
暨QuantumATK Workshop 2018
由Synopsys QuantumATK、山东师范大学物理与电子科学学院与费米科技(北京)有限公司共同主办的“材料与器件模拟研讨会暨QuantumATK Workshop 2018”,将于2018年10月12-14日在山东省济南市山东师范大学举办。 随着二维材料等低维功能材料的涌现,新型电子器件的设计和模拟正在扮演者越来越重要的角色。为了共同探讨“材料与器件”领域的学术问题,我们将邀请QuantumATK的资深用户分享交流研究经验,同时也热忱欢迎广大老师和同学在会上介绍自己的最新进展工作。届时,Synopsys QuantumATK公司的 Dr. Kurt Stokbro 与 Dr. Marcus Yee 也将为广大 ATK 用户与研究者详解 QuantumATK 2018 版最新功能,介绍相关领域的最新进展。 会议主题 材料与器件模拟专题讲座 用户报告 QuantumATK 2018年新版软件上机练习 交流与答疑 详情请期待后续更新。 会议详情 日期: 2018年10月12-14日 地点: 山东师范大学长清湖校区(山东省济南市长清大学城大学路1号) 费用: 免费(食宿交通等费用自理) 人数:80人 报名表见文末 日程安排 12日:早上报到 12-13日:专题讲座与用户报告 13-14日:上机操作与答疑 会前准备 自带笔记本电脑 提前到https://eval.synopsys.com申请软件试用获取license(必须是学术邮箱申请,请注明China Workshop) 酒店推荐 (酒店房间紧张,请尽早预定) 如家快捷酒店(济南长清大学城店) 长清大学城三庆青年城1号楼; 电话:(0531)55760888 都市118连锁酒店 济南市长清区大学城商业街5号楼(质诺工坊北邻四楼); 电话(0531)55612888 格林豪泰快捷酒店(济南长清大学城店) 长清大学城商业街3-1号; 电话:(0531)87310999 […]
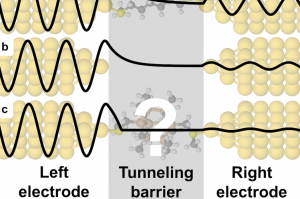
【QuantumATK亮点文章】研究一种极端的分子电子绝缘体
最近 Nature[1]上发表了一项研究,提出了一种新型的分子结,这种分子结比同样尺度的真空区域更加“绝缘”。这个效应是通过设计分子实现量子相消干涉实现的,这些分子在某些能量范围里强烈的抑制电子在分子中的透射。 输运性质的计算是由哥本哈根大学的Gemma Solomon组使用QuantumATK完成的。 合成与电导的实验测量是由上海师范大学的肖胜雄、哥伦比亚大学的Colin Nukolls和Latha Venkataraman等研究组合作完成的。这类分子可能用在热电或其他电子器件中作为绝缘体。 图1. 电子透过单分子结隧穿时的波函数衰减示意图。(a)透过低导电性分子(烷链)时;(b)透过空结(即电极间是真空);(c)透过一个分子发生量子相消干涉的情况,隧穿概率非指数衰减。 参考资料 所有文中所涉及的计算方法均在QuantumATK中提供,详见以下教程: 相关的中文教程列表 英文教程 Four tutorials on molecular electronics Tutorial on studying the electron transport properties of a graphene nanoribbon with a distortion 参考文献 [1] M. H. Garner, H. Li, Y. Chen, T. A. Su, Z. Shangguan, D. W. Paley, T. Liu, F. Ng, H. […]
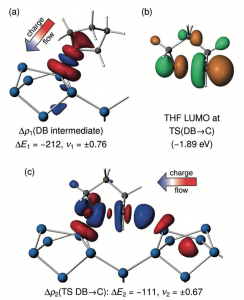
BAND Highlight:硅表面醚吸附-计算表面化学与分子计算化学共同点的最佳案例(Angew. Chem. Int. Ed., 2017)
文献资料:Lisa Pecher, Slimane Laref, Marc Raupach, and Ralf Tonner, Ethers on Si(001): A Prime Example for the Common Ground between Surface Science and Molecular Organic Chemistry, Angew. Chem. Int. Ed. 2017, 56, 15150 –15154 通过计算化学研究表明,在超高真空条件下,醚分子在Si(001)表面的吸附可以用有机化学的经典概念来理解。两步反应机理的详细分析:1)醚的氧原子与Lewis酸性表面原子之间形成的配价键(DB);2)附近Lewis碱性表面原子的亲核攻击表明,它反映了溶液中酸催化的醚裂解。 O-Si键是这类键中最强的,并且第2步的反应活性违背了Bell-Evans-Polanyi原理。本文使用一种新的键分析方法(pEDA-NOCV),对C-O键解离过程中的电子重排进行了可视化的研究。 结果表明,半导体表面亲核取代的机理与Sn2分子反应的机理是一致的。我们的发现表明了表面科学和分子化学如何相互受益,并得到意想不到的洞察方式。 BAND,是目前最先进的专注于材料化学计算研究工具。
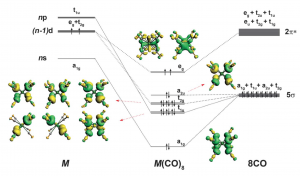
ADF Highlight:钙锶钡配合物也遵守18电子规则(Science, 2018)
文献资料:Xuan Wu, Lili Zhao, Jiaye Jin, Sudip Pan, Wei Li, Xiaoyang Jin, Guanjun Wang, Mingfei Zhou, Gernot Frenking, Observation of alkaline earth complexes M(CO)8 (M = Ca, Sr, or Ba) that mimic transition metals, Science 361, 912–916 (2018) 碱土金属钙(Ca)、锶(Sr)和钡(Ba)主要通过nS和nP价轨道进行化学键合(其中n为主量子数)。复旦大学周鸣飞老师课题组和南京工业大学Gernot Frenking与赵莉莉老师课题组报道了八配位羰基配合物M(CO)8(其中M=Ca,Sr或Ba)在低温氖基质中的分离和光谱表征。对这些立方Oh对称配合物的电子结构分析表明,M-CO键主要来自[M(dπ)]→(CO)8 π的反馈作用,从而解释了C-O伸缩频率的强烈红移。还制备了相应的自由基阳离子气相配合物,并通过质量选择红外光解光谱对配合物进行了表征,证实其遵守过渡金属化学有关的18电子规则。 本文使用了一种非常强大的研究化学成键细节的工具:ADF中的EDA-NOCV功能(在BAND模块中,该功能对周期性体系同样适用)。 EDA功能将金属与配体之间的相互作用能,分解为泡利排斥、静电作用、轨道作用,并得到分子轨道与碎片轨道之间的关系,而NOCV功能将轨道作用更详细地分解到具体的一对对相互作用的轨道中。 NOCV分析表明,M-CO键主要来自[M(dπ)]→(CO)8 π的反馈作用: 相关中文教程: ETS-NOCV功能案例:开壳层、闭壳层、环状结构 ETS-NOCV功能案例:二聚体能量分解(EDA)中轨道作用、电子在片段轨道间转移的分解 键能分解(EDA) NOCV理论
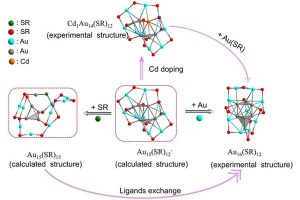
ADF Highlight:Au16(S-Adm)12与Cd1Au14(StBu)12的全结构确定以及Au15(SR)13结构的应用(JACS, 2018)
文献资料: Sha Yang, Shuang Chen, Lin Xiong, Chong Liu, Haizhu Yu, Shuxin Wang, Nathaniel L Rosi, Yong Pei, and Manzhou Zhu, Total Structure Determination of Au16(S-Adm)12 and Cd1Au14(StBu)12 and Implications for the Structure of Au15(SR)13, J. Am. Chem. Soc., Just Accepted Manuscript • DOI: 10.1021/jacs.8b04257 纳米团簇(例如Au15(SR)13)不但在生物领域的应用至关重要,也是理解金配合物向金纳米团簇转变机理的关键。然而这些过渡尺寸金纳米团簇的确定,一直以来是一个重大挑战。 本文得到了两个新的过渡区纳米团簇,包括迄今为止最小的合金纳米团簇Cd1Au14(StBu)12,以及同晶纳米团簇Au16(S-Adm)12,并通过单晶X射线衍射确定了它们的原子结构。此外,基于Cd1Au14(SR)12和Au16(SR)12的结构,湘潭大学裴勇老师课题组进行了DFT计算,预测了“转变”纳米团簇Au15的结构(Au15(SR)12–和Au15(SR)13)。这项工作,搭建了金配合物与金团簇之间桥梁。

ADF Highlight:两个莫比乌斯共轭纳米环组成的可分离环烷(Nature Comm.,2018)
文献资料: Yang-Yang Fan, Dandan Chen, Ze-Ao Huang, Jun Zhu, Chen-Ho Tung, Li-Zhu Wu & Huan Cong, An isolable catenane consisting of two Möbius conjugated nanohoops, Nature Communications, 9, 3037 (2018) 莫比乌斯拓扑,除了数学上的重要性外,在分子水平上也很吸引人,结构优雅而独特。然而合成却颇具挑战性。尽管一些莫比乌斯型分子已被化学家合成、分离,并进行了广泛的理论计算研究,但稳定的莫比乌斯共轭分子的设计、制备和表征至今仍是一项重要的任务,更不用说将分子莫比乌斯带组装成更复杂的拓扑了。 本文报道了由两个完全共轭的纳米莫比乌斯环组成的有机环烷的有效合成、晶体结构和理论研究结果。这项工作强调了寡对苯撑(oligoparaphenylene)衍生的纳米环(高度扭曲、合成具挑战性的共轭大环),不仅可以作为互锁超分子结构的构建块;而且还代表新的化合物种类——通过非共价相互作用稳定的,可分离的莫比乌斯构象。 本文使用ADF2017.104,在M06-2X泛函/DZP基组水平上,进行了共轭环间的相互作用能分解分析,由中科院大学丛欢课题组、厦门大学朱军课题组共同完成。 软件相关功能中文教程: 如何进行片段之间相互作用能、分子之间相互作用能、键能、键解离能、结合能计算、键能分解EDA、片段轨道布居 ETS-NOCV功能案例:二聚体能量分解(EDA)中轨道作用、电子在片段轨道间转移的分解 非键作用NCI: Non-Covalent Interactions

QuantumATK在半导体和微电子工业研究中的应用
英特尔:铜纳米线的电子输运特性 文章(PHYSICAL REVIEW B 92, 115413 (2015))使用密度泛函理论以及密度泛函紧束缚近似模型,对直径约 1 nm 和 3 nm 的铜纳米线的不同晶向和表面端基原子的电子输运性质进行了研究。发现无论何种直径、晶向、端基原子均不影响其金属性。电子透射强烈的依赖于晶向和端基原子,沿 [110] 方向的铜纳米线透射最强。与无端面的纳米线相比,表面氧化纳米线的电子透射显著降低。 详细介绍:http://www.fermitech.com.cn/vnl-atk/semi_industry_001/ 美光:绝缘体中的金属杂质的影响与阻变存储器 文章(JOURNAL OF APPLIED PHYSICS 117, 054504 (2015))对铜掺杂SiO2和Al2O3在热力学、动力学和电子性质方面进行了原子尺度的模拟研究,得到不同浓度(9.91×1020 cm-3和3.41×1022 cm-3)的铜掺杂下的定量结果。金属-绝缘体界面导致体系的形成能与块体相比降低大约4eV。另外,本文还介绍了Cu-Cu相互作用对降低化学势的重要性。这些概念的讨论是在关于阻变存储器(RRAM-M)局域传导路径的形成及其稳定性的背景下展开的。电子态密度以及通过这些局域路径的非平衡透射研究确认了透射增大了三个数量级。本文通过原子尺度的漂移-扩散计算,研究了这些传导路径的动力学行为。本文最后对RRAM-M的原胞进行了分子动力学模拟,试图将上述所有现象用统一的自洽的模型结合起来。 详细介绍:http://www.fermitech.com.cn/vnl-atk/semi_industry_002/ 松下:金属-有机体系的载流子注入 文章(Surface Science 600 (2006) 5080–5083)研究了单个π共轭分子吸附或夹在两个电极上的电流特性,尤其是载流子通过有机/金属界面的注入。首次使用第一性原理方法研究了分子的取向和电极材料对电流对影响:过去的电流计算都是假设分子与电极之间通过共价键连接。 详细介绍:http://www.fermitech.com.cn/vnl-atk/semi_industry_003/ 三星:金属-半导体界面接触电阻 文章(APPLIED PHYSICS LETTERS 105, 053511 (2014))使用密度泛函理论和密度泛函紧束缚近似研究了n-Si在[100]方向电阻率下限的理想金属电子结构效应。结果表明,在高掺杂浓度时,“理想金属”假设在某些情况下会失效,因此对n-Si的接触电阻下限至少低估了一个数量级。金属和半导体在横向动量空间的失配,也就是所谓的“谷过滤效应”,对使用的原胞的横向边界情况的细节非常敏感。因此在金属-半导体接触面的电子输运的原子尺度模拟,需要明确所包含的金属原子和电子结构。 详细介绍:http://www.fermitech.com.cn/vnl-atk/semi_industry_004/ 格罗方德:肖特基势垒的调制 文章使用密度泛函理论研究了肖特基势垒高度(Schottky Barrier Height)对界面形态的依赖,以及如何通过在 NiSi2/Si界面替位掺杂进行调制。使用 meta-GGA 交换相关泛函预测出相当精确的Si带隙。本文的研究表明,(n型半导体的)电子肖特基势垒高度在(001)方向显著低于(111)方向。这些结果定性上与界面形态依赖的肖特基势垒高度的实验结果一致。肖特基势垒高度能够通过 NiSi2/Si 界面替代掺杂显著降低,本文讨论了 Si 掺杂位点、杂质类型以及晶向对优化肖特基势垒高度的影响。 详细介绍:http://www.fermitech.com.cn/vnl-atk/semi_industry_005/ IBM:纳米线作为下一代晶体管互连技术的可能性 […]
DFT-PlaneWave:QuantumATK中的平面波密度泛函理论计算引擎
概述 QuantumATK 材料与器件模拟平台包含的 DFT-PlaneWave 全功能平面波密度泛函理论计算引擎与平台的图形用户界面 NanoLab 完美集成,可能是目前最灵活和友好的平面波程序。DFT-PlaneWave 实现了赝势和平面波基组相结合的第一性原理密度泛函理论电子结构计算方法。DFT-PlaneWave 使用内置的模守恒赝势,涵盖了元素周期表中绝大部分的元素。ATK-DFT 实现了众多版本的局域密度近似(LDA)和广义梯度近似(GGA)的交换关联函数。meta-GGA、DFT-1/2、HSE06杂化泛函等则可以准确、快速地计算半导体、绝缘体材料的带隙。 更多功能介绍详见 QuantumATK功能列表。 基于平面波基组的 DFT 计算引擎 完全自主开发的代码,完美集成于图形界面环境 针对所有元素都有默认的网格截断能设置支持杂化泛函(使用新颖的 Adaptively Compressed Exchange(ACE)算符方法,比传统方计算性能更佳) 参考:ArXiv 或 ACS.JCTC 可选模守恒 Troullier-Martins 赝势 FHI/SG15/PseudoDojo势,用于周期表几乎全部元素,很多元素支持半芯势PseudoDojo 和 SG15 势是全相对论的 可选Projector Augmented Wave(PAW)势 使用更小的平面波截断能计算GPAW/JTH两套PAW势数据,支持稀土元素计算 超过 300 种 LDA/GGA/metaGGA 交换相关泛函(libXC) LDA:HL,PW,PZ,RPA,WIGNER,XA 等GGA:BLYP,BP86,BPW91,PBE,PBES,PW91,RPBE,XLYP 等metaGGA:SCAN多种杂化泛函HSE06、B3LYP、B3LYP5等供选择用于半导体和绝缘体精确带隙计算的方法 MetaGGADFT+1/2经验的“赝势投影算符移动”(Pseudopotential Projector Shift,PPS)方法(内含 Si 和 Ge 的参数) 范德华力模型(DFT-D2 和 DFT-D3)非共线、限制性和非限制性的自旋极化计算自旋-轨道耦合本征值求解 默认使用 Generalized Davidson method – 稳定强壮的方法还包含 […]
QuantumATK O-2018.06 新版发布
这是自 2017 年 9 月加入 Synopsys 以来的第一次发布,从这一版本开始,ATK 更名为 QuantumATK。 QuantumATK 材料与器件模拟平台包含量子力学方法(DFT 和半经验模型)和以下几个模块:非平衡态格林函数(NEGF),经验力场(ForceField) 和图形用户界面(NanoLab,即过去的 VNL)。 此版本包含了大量的 NanoLab 功能更新,进一步提升了周期体系的 DFT 和 DFT-NEGF 的计算性能,正式发布了 PlaneWave 计算引擎(含 HSE06)。全新的 Study Object 框架可以用于进行非常复杂的计算自动化,比如器件体系的结构优化,获取伏安特性,模拟电中性或带电的点缺陷,等等。此外,QuantumATK 还优化了默认参数取值,提高了计算精度。 License 和下载方法 如果您是用户,您可以从 SolvNet网站 直接下载最新版本。 (如果您还没有注册 SolvNet,请尽快注册。注册时需要提供用于识别用户的 Synopsys Site ID,如果您不知道Site ID,可以与我们联系。) 如果您还不是用户,但是想尝试一下 QuantumATK,您可以通过 Synopsys EVAL 网站申请30天的试用 license。 更新概要 平面波PlaneWave DFT 代码 全新赝势 全新的半经验模型 全新的力场模型 大量的计算性能提升 全新的 Study Object […]