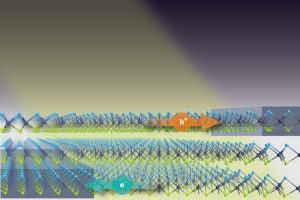
之前的文章(链接)介绍了在原子尺度上模拟光电池器件时考虑温度效应的方法[1],最近这种方法又被用于一种新的堆叠 Janus 光电池器件[2]。基于最新发现的二维材料MoSSe的超薄(0.5-1nm)器件可以产生的光电流和外量子效率(EQE)比 20-40 倍厚的硅基器件还要大。 这类 Janus 过渡金属二硫族化合物(TMD)是一种双侧不对称材料,跨平面的不对称性在二维材料平面的两侧产生了一个偶极,这个偶极可以多层堆叠。这样得到的“p–n结”可以用于分离在底层和顶层产生的电子(e-)和空穴(h+)载流子,载流子分别进入两侧的石墨烯电极中产生光电流。至关重要的是,石墨烯不会像金属电极一样屏蔽这种跨层的偶极。 使用 QuantumATK 的图形界面可以基于 TMD 创建堆叠Janus光电池器件。QuantumATK的第一原理DFT和DFT-NEGF 方法、光电流计算模块等可以用于计算能带、态密度、电子透射、输运通道、光电流密度等各种重要性质。文章报道的 Janus 光电池器件可以产生的光电流和外量子效率(EQE)比 20-40 倍厚的硅基器件还要大。此外作者还注意到,由于偶极的堆叠影响,器件在光子能量小于单层 Janus MoSSe 的带隙时也能产生光电流。作者建议也许可以使用 MoSSe Janus 层与硅薄膜结合来提高硅对低能量光子的吸收效果。作者还建议可以在光电应用领域里研究其他 Janus 二维材料(例如 CrSSe、ZrSSe 等)。 相关教程和讲座 文章中所用方法都在QuantumATK O-2018.06之后的版本实现,详见: Webinar on accurate atomistic simulations of solar-cell devices including temperature effects Tutorial on the photocurrent in a silicon p-n junction Tutorial on electron transport […]