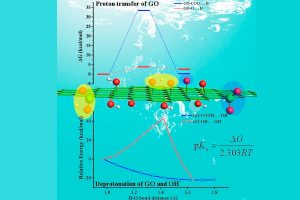
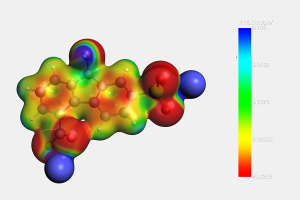
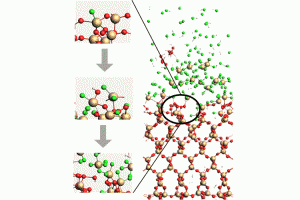
在水相环境中,了解纳米氧化石墨烯(GO)的表面酸性特征以及含氧官能团的相关质子转移行为,对于氧化石墨烯纳米材料的应用具有重要理论意义和实际价值。南京工业大学杨晓宁教授课题组,采用密度泛函(DFT)方法研究了GO的酸性和表面含氧基团的质子转移行为。DFT计算使用AMS软件中的ADF模块,采用了B3LYP泛函和经验色散校正(-D3)。 在模拟中,隐式COSMO溶剂化模型用来考虑水的溶剂化效应。研究首先设计提出模拟计算了GO的羧基和羟基的酸离解离常数(pKa)的热力学方法,模拟结果与实验数据呈现一致性,模拟结果明确了GO的每种类型官能团的酸性常数数值大小,也为相关纳米颗粒体系酸性参数的理论研究提供了方法参考。通过热力学和动力学计算揭示了GO上羧基基团解离是其表面电荷形成的主要来源。进一步的DFT计算表明质子很容易在相邻的羟基和环氧基团之间转移。在质子转移过程中,GO表面的电荷分布发生显著变化。研究展现了表面含氧基团间的面内质子转移在GO表面电荷调控中的重要性,结果也将有助于加深对GOs表面化学行为新的认知。 参考文献: Yushuang Lu, Lijuan Huang, Yanan Guo, Xiaoning Yang, Theoretical insights into origin of graphene oxide acidity and relating behavior of oxygen-containing groups in water, Carbon, Volume 183, 355-361