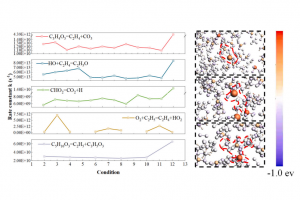
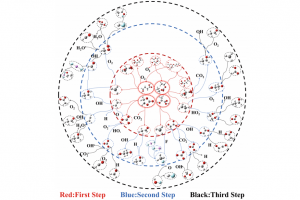
研究背景 锂离子电池因高能量密度被广泛应用,但其热失控问题日益突出,严重威胁使用安全。热失控过程涉及电解液分解、链式反应及气体释放等多阶段复杂反应,不同电解质体系和锂盐添加剂对热行为的影响机制尚不明确。传统实验方法难以揭示微观反应路径和自由基演化规律。因此,亟需结合实验与模拟手段,系统研究电解质组成及 LiPF6 浓度对热失控的调控机制,为提升电池热安全管理提供理论依据。 研究方法 本研究选取三种不同电解液体系的软包磷酸铁锂电池,通过加速量热仪进行 3C 过充实验,分析热失控过程中的温度分布和气体组成。采用 ReaxFF-MD 模拟不同 EC/DEC/DMC 配比及LiPF6浓度(0%、5%、10%)下的电解液燃烧过程,追踪反应物、中间产物及自由基演化,识别关键基元反应路径。结合 DFT 计算电解液分子及络合物的静电势、前线轨道能级和弱相互作用,从电子层面揭示不同组分对热失控链反应的调控机制。 图1 电解质分子反应系综 图2 DFT 分析:(a) 不同电解质与 LiPF6 的静电势;(b) 不同电解质、LiPF6 及其配合物的 HOMO-LUMO 轨道;(c) 关键基元反应的反应速率;(d) 分子反应动力学中关键基元反应的电荷分布;(e) 配合物的 IRI 弱相互作用 主要研究结论 本研究揭示了不同电解质体系及 LiPF6 浓度对锂离子电池热失控行为的调控机制。实验结果表明, LF653465 电池热失控发展最快(仅 600.5 s 达到峰值温度),而 LF5820117 电池热稳定性相对较高。气体分析显示,热失控后主要产生 CO、CO2、H2、CH4、C2H4 等气体,其比例差异反映了不同溶剂的分解路径特征。 基于 AMS 软件的 ReaxFF-MD 模拟发现,纯 EC 体系主要通过碳酸酯环开环引发链反应(R4: C3H4O3 = C2H4 + CO3),能垒为 0.1637 […]