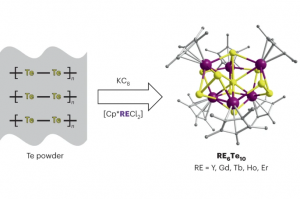
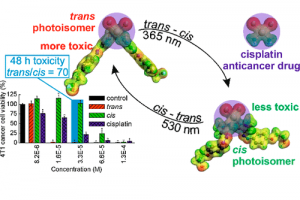
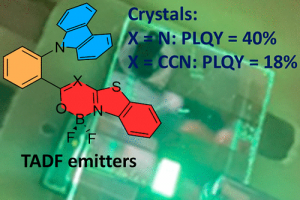
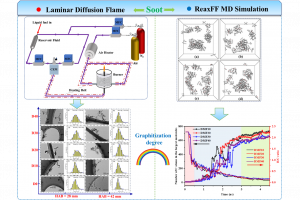
锎化合物的慢磁弛豫 本文报道了大环锎衍生物 Na[Cf(H2O)(DOTA)] (DOTA = 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetate), 1-Cf 的合成和表征,并将其与镝对应物 Na[Dy(H2O)(DOTA)], 1-Dy 进行了比较研究。在 1-Cf 和 1-Dy 之间观察到发散的光谱和磁行为。根据光谱测量,作者提出可及的 5f→6d 跃迁(可能与电荷转移跃迁协同工作)是观察到的 1-Cf 宽带光致发光的主要因素。1-Cf 的直流磁化率数据显示,与之前观察到的 1-Dy 相比,1-Cf 的磁矩较低。计算表明,游离 f9 离子是更大配体场效应的结果。 值得注意的是,1-Cf 在交流磁化率测量的时间尺度上显示出缓慢的磁弛豫,使其成为基于锎的单分子磁体的第一个例子。交流磁化率数据的并排比较揭示了 1-Cf 和 1-Dy 之间的磁弛豫特性差异很大。这种发散弛豫行为主要归因于 Dy3+ 和 Cf3+ 之间自旋轨道耦合的固有差异。 Slow Magnetic Relaxation in a Californium Complex, Luis M. Aguirre Quintana etc, J. Am. Chem. Soc. 2024, DOI: 10.1021/jacs.4c10065 下一代三价锕系元素和镧系元素分离萃取剂的计算辅助开发(四川大学) 三价锕系元素 [An(III)] 和镧系元素 [Ln(III)] 之间的化学相似性,对它们的区分和分离提出了重大挑战,这是关闭核燃料循环的关键一步。然而,现有的分离方法通常存在分离因子不足、汽提效率有限和不希望的共萃取等缺点。 […]