Amsterdam Modeling Suite(简称AMS,原名ADF),是一款更专业的材料化学模拟平台。可以在原子水平研究分子、溶液、固体表面反应、吸附结构、聚合物的电子态结构、化学反应、谱学性质、化学键性质、光电性能、热力学性质、力学性质、电磁学性质等。 AMS 是调用ADF、BAND、MOPAC、DFTB、ReaxFF、ForceField完成计算的驱动程序,可以调用这些子模块完成微观动力学、混合模拟、分子动力学模拟、巨正则系综蒙特卡洛模拟。 微观反应动力学:Microkinetics方法介绍(点击),案例教程(点击) 混合计算: 轻松地通过分区,实现DFT、MM、ReaxFF、DFTB等任意混合,分区数量没有限制,该功能支持周期性体系,参考:Hybrid调用多种计算引擎分区计算 混合计算涉及MM的力场包括:UFF、GAFF、Amber、Tripos GCMC模拟:ADF-GCMC、BAND-GCMC、ReaxFF-GCMC、DFTB-GCMC、MOPAC-GCMC…… 分子动力学模拟(以下算法均适用于ADF-MD、BAND-MD、DFTB-MD、MOPAC-MD、ReaxFF-MD) REMD加速反应算法,用法参考:新的分子动力学反应加速算法REMD Bond Boost加速反应算法,用法参考:ReaxFF-Bond Boost:加速分子动力学模拟中反应发生 CVHD加速反应算法 Molecule Gun(入射分子)、Molecule Sink(定时移除指定组分)功能 扩散系数、自相关函数 ADF模块 大体系计算,例如大体系吸收光谱 过渡金属、重元素体系 最先进的相对论方法,计算自旋-轨道耦合 丰富的谱学性质、非线性光学、热力学、核磁共振、电子自旋共振等 成键分析、电荷与电子密度分析 最新泛函,例如SCAN、-D4(EEQ)色散修正泛函、丰富的LibXC泛函、高精度双杂化泛函 分子间相互作用精确计算MP2方法 图形界面简单方便,初学者也能很好使用 特色功能: 成键分析与化学反应:能量分解EDA、电荷分解CDA、化学价自然轨道ETS-NOCV、分子轨道MO投影到碎片轨道SFO、键级、通过Laplacian电子密度与键临界点区分化学键类型、DORI、过渡态搜索 光学:荧光、磷光辐射跃迁寿命、磷光发射谱、荧光发射谱、SOCME估算系间窜跃、紫外可见吸收谱(非相对论方法、相对论动能修正、考虑自旋轨道耦合)、红外光谱、拉曼光谱、表面增强拉曼光谱、Nucleus Independent Chemical Shifts(NICS,核独立化学位移)的计算、X射线吸收谱(XANES、EXAFS、XPS)、VCD、MCD、ESR、EPR、零场劈裂ZFS、Frank-Condon谱、极化率、穆斯保尔谱、旋轨耦合(SOCME)、POLTDDFT方法快速计算Au、Ag团簇吸附小分子体系紫外可见吸收谱 非线性光学 :超极化率、衰减一阶超极化率β、衰减二阶超极化率γ、零频β、光学矫正β、EOEP β、SHG β、零频γ、EFIOR γ、OKE γ、IDRI γ、EFISHG γ、THG γ、TPA γ 其他特殊理论方法:FDE方法、收缩变分DFT(CV(n)-DFT)用于单重态-三重态激发的计算(该功能不像普通的TDDFT那样被电荷转移激发所困扰)、配体场DFT(LFDFT)(对 d → d和f → d电子转移的情况,令计算结果更可靠)、微扰局域分子轨道 电荷、电子密度分析:AIM(Bader)、Natural Population Analysis(NPA)、Mulliken电荷(任何计算完毕即可在Output或View中查看)、Hirshfeld电荷(任何计算完毕即可在Output或View中查看)、Voronoi形变电荷(任何计算完毕即可在Output或View中查看)、ELF、NCI、SEDD(默认单点计算完毕即可在View中查看)、DORI(默认单点计算完毕即可在View中查看)、RGD(默认单点计算完毕即可在View中查看)、基态差分电子密度、电子激发差分电子密度 电荷迁移性:转移积分方法计算分子间载流子迁移性、金属-配体电荷转移(MLCT)、激发态电荷转移描述符 溶剂化:COSMO、SCRF、3D-RISM、FDE […]


神经毒剂模拟物甲基对氧磷在光活性纳米织物上选择性可见光驱动毒性降解(Appl Catal B-Environ 2020)
本文亮点: 利用静电纺丝技术制备了可见光催化纳米织物 这种纳米织物是以铌酸铁和聚己内酯为基底 结果表明,它对甲基对氧磷具有极强的解毒选择性 光生H+和•OH自由基是导致神经毒剂破坏的原因 通过DFT进行理论计算证实了纳米织物的效率 巴西米纳斯吉拉斯联邦大学化学系、物理系、冶金与材料工程系、理工学院多个课题组联合研究,以聚己内酯(PCL)和固化铌酸铁(NbOFe)纳米粒子为基底,采用静电纺丝技术制备了一种高效的光催化纳米织物(NbOFe-NF),并将其应用于高神经毒性化学战剂甲基对氧磷(MP)的光降解。其中光催化试验没有任何溶剂参与,仅仅依赖于织物、基底和可见光辐射。结果表明,光催化48小时,MP的转化率为94.5%。此外,还发现光活性纳米织物具有极强的选择性,可将MP及其原始有机磷产品转化为毒性较小的化合物。整个过程完全是光催化的,通过环境湿度产生H+和•OH自由基。 理论研究中,使用AMS-BAND进行计算,电子轨道使用未收缩的STO基组TZ2P展开,该基组具有三重ζ,并为每个原子增加了两组极化函数,并使用ZORA方法考虑相对论效应。几何结构优化和能量计算GGA-OLYP获得,对MP在铌酸铁表面吸附的能量分布进行了计算研究。为了模拟非晶态结构,在材料结构中保留了与氢结合的氧原子。在AMS中建立了(001)表面的平板模型,并创建超胞,游离MP分子在相应的表面进行了结构优化,仅吸附于平板的一侧。BAND在二维、一维材料表面吸附计算的精度与效率,均高于平面波方法。 参考文献: Selective visible-light-driven toxicity breakdown of nerve agent simulant methyl paraoxon over a photoactive nanofabric, Applied Catalysis B: Environmental, Available online 9 December 2020, 119774
AMS2020发布
AMS2020对软件整体进行了重构,各模块的整合协同性进一步提高,以AMS引擎统一驱动各个模块。同时也有一些功能上的进展: ADF模块 数值频率、PES等计算,在原先MPI并行的基础上,增加驱动层面的并行,也就是说,可以同时计算多个结构点,而不需要像过去的版本,一个一个点顺序计算(实现方式) 快速G0W0、RPA单点计算 对称性的默认设置改变为Nosymm 增加了元素Uue(Z=119)、Ubn(Z=120)的基组 默认使用Scalar相对论 ADF via AMS整合进入ADF 电荷转移积分,(近)简并能级将会一起得到处理。 BAND模块 增加了元素Uue(Z=119)、Ubn(Z=120)的基组 默认使用Scalar相对论 计算指定k点有效质量 DFTB模块 方法包括DFTB、SCC-DFTB、DFTB3、GFN1-xTB、NonSCC-GFN1-xTB。对多k点体系,GFN1-xTB计算速度大大提高 Machine Learning Potentials 这是一个经验性的第一性原理计算模块 AMS和多个ML Potentials后端之间的接口 预优化按钮增加了ML Potentials中的ANI-1cxx方法 多层计算 轻松地通过分区,实现DFT、MM、ReaxFF、DFTB等任意混合,分区数量没有限制,该功能支持周期性体系。 新的力场包括:UFF、GAFF、Amber、Tripos ReaxFF ReaxFF与ReaxAMS合并,统一使用AMS驱动 支持Bulk(三维)、Slab(二维)、Chain(一维)、None(非)周期边界条件 COSMO-RS 质子化和聚集态:脚本工具处理不同流体相中不同状态,以提高溶解度、pKa和其他热力学预测 分析功能 自相关函数,扩散系数 低频热力学修正
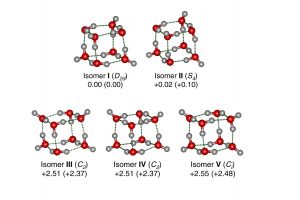
最小冰粒氢键拓扑结构的红外光谱研究(Nat. Comm. 2020)
水八聚体的立方结构由六个四元环组成的团簇体系,能很好的用来解释氢键拓扑结构细微变化所驱动的协同作用。虽然许多不同的结构被预测出来,但从振动光谱中提取出结构信息仍待实现,这需要电中性团簇的尺寸选择性具有足够的分辨率来识别不同异构体的贡献。清华大学/南方科技大学李隽课题组、胡撼石课题组、中国科学院大连化学物理研究所杨学明课题组、张东辉课题组和江凌课题组报导了使用可调真空紫外自由电子激光器阈值光离子化方案,测得孤立的冷冻、电中性八聚水的特定尺寸红外光谱,结果观察到大量的尖锐振动带特征。 对红外光谱的理论分析表明存在五个立方异构体,其中两个具有手性。这些结构的相对能量反映出不同的拓扑相关、离域多中心氢键作用。这些结果表明,即使有共同的结构特征,氢键网络之间的合作程度差异也导致了不同层次的结构。 为了解水八聚体的电子结构,作者利用离域定域分子轨道(LMO)理论分析了立方异构体的氢键网络,进行了自然键轨道(NBO)、自适应自然密度分配(AdNDP)、能量分解分析-化学价自然轨道(EDA-NOCV)和主相互作用轨道(PIO)等分析。 利用AMS软件ADF模块,在GGA-PBE/TZ2P水平上,利用EDA-NOCV分析了立方异构体中氢键相互作用的的本质。EDA-NOCV方案提供了关于化学键中轨道相互作用强度和贡献的定性(Δρorb)和定量(ΔEorb)信息。 参考文献: Gang Li, Yang-Yang Zhang, Qinming Li, Chong Wang, Yong Yu, Bingbing Zhang, Han-Shi Hu, Weiqing Zhang, Dongxu Dai, Guorong Wu, Dong H. Zhang, Jun Li, Xueming Yang & Ling Jiang, Infrared spectroscopic study of hydrogen bonding topologies in the smallest ice cube, Nature Communications volume 11, Article number: 5449 (2020)
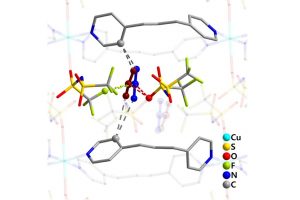
双(三氟甲磺酰基)酰亚胺阴离子与被吸附二氧化碳之间相互作用(Communications Chemistry 2020)
离子液体(ILs)的二氧化碳(CO2)选择性吸收特性与CO2捕集方法的发展密切相关。尽管有报道称氟化组分使ILs增强了CO2溶解度,但深入理解ILs与CO2之间的相互作用一直是一个挑战。在本研究中,作者利用软晶质材料[Cu(NTf2)2(bpp)2] (NTf2‒ = bis(trifluoromethylsulfonyl)imide, bpp = 1,3-bis-(4-pyridyl)propane)作为单晶X射线衍射分析的替代物,将CO2与NTf2‒(氟化离子液体组分,导致二氧化碳高溶解度)之间的相互作用可视化。对负载二氧化碳的晶体结构的分析表明,CO2与NTf2‒阴离子的氟原子和氧原子以反式而非顺式结构发生相互作用。对负载CO2的晶体结构的理论分析表明,CO2与骨架之间存在色散和静电相互作用。总而言之,为理解和改进离子液体吸收二氧化碳的特性提供了重要的见解。 使用AMS-ADF优化添加H原子的结构(PBE-D3(BJ)/TZ2P),并使用能量分解方法(EDA)结合化学价态理论的自然轨道对模型(NOCV)结构进行了分析。 参考文献: Xin Zheng, Katsuo Fukuhara, Yuh Hijikata, Jenny Pirillo, Hiroyasu Sato, Kiyonori Takahashi, Shin-ichiro Noro & Takayoshi Nakamura, Understanding the interactions between the bis(trifluoromethylsulfonyl)imide anion and absorbed CO2 using X-ray diffraction analysis of a soft crystal surrogate, Communications Chemistry volume 3, Article number: 143 (2020)
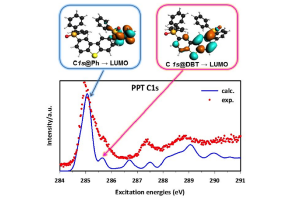
OLEDs双极性磷光基质材料的XPS和NEXAFS研究
单通道的Kohn-Sham DFT理论结合跃迁势(Transition Potential),能够考虑核空穴形成的大部分的电子弛豫效应,从而描述轻原子的K-壳层NEXAFS光谱。这种方法提供了一组正交轨道,从中可以得到跃迁偶极矩。 K壳层NEXAFS光谱是通过对每个非等效原子位的激发光谱进行单独计算,并将其贡献按相对权重相加得到的。这样就可以将总的光谱性质反卷积到不同组分中,从而有助于将光谱特征分解到分子的特定部位。 这种方法,最近被应用到2,8-bis-(diphenylphosphoryl)-dibenzo[b,d]thiophene(PPT)的C1s和O1s NEXAFS光谱的模拟,这是最近引入OLED中的一种双极性磷光主体材料,用于解释在Trieste的电子同步加速器气相束线处获得的实验光谱。PPT可以认为是由两个二苯基氧化膦(dPPO)部分,对小二苯并噻吩(DBT)核心官能化而形成。 在C的K-边的DFT-TP计算表明,PPT的C1s谱主峰归属于dPPO臂的苯环,而第二弱峰则归属于PPT DBT核的苯环部分。 本研究的结论对OLED的未来应用具有重要意义:PPT的氧化膦基团是PPT的DBT核与外层基团之间π共轭的断裂点。然而,这些基团在很大程度上不影响DBT中心部分的电子性质。 参考文献: A. Guarnaccio, T. Zhang, C. Grazioli, F. Johansson, M. Coreno, M. de Simone, G. Fronzoni, D. Toffoli, E. Bernes, C. Puglia, PPT Isolated Molecule and Its Building Block Moieties Studied by C 1s and O 1s Gas Phase X-ray Photoelectron and Photoabsorption Spectroscopies, J. Phys. Chem. C […]
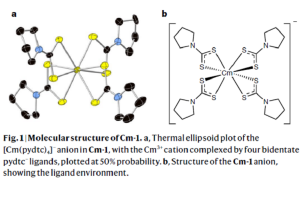
压缩吡咯烷二硫代氨基甲酸锔提高共价性(Nature. 2020)
锔在锕系元素中是独一无二的,因为它的半填充5f7壳层比其他5fn结构的能量更低,因此既难于氧化还原,(5f壳层)又不易形成化学键。这一点对钆(钆是镧系中锔的类似物)更为明显,因为相对于锔的5f轨道,钆的4f轨道更为紧缩。 然而在高压下,锔的5f电子从局域化状态转变为巡游态。这种转变形成一种晶体结构,这种结构由锔原子之间的磁相互作用决定。那么是否也可以通过施加压力来改变锔(III)-配体相互作用中的前沿金属轨道,从而诱导形成具有一定共价性的金属-配体键? 弗洛里达州立大学Thomas E. Albrecht-Schönzart,纽约州立大学Eva Zurek、Jochen Autschbach,亚琛工业大学Manfred Speldrich等课题组合作,报道了在高压(高达11GPa)下,[Cm(pydtc)4]–(pydtc,吡咯烷二硫代氨基甲酸基)的锔-硫键中,5f/6d轨道角色变化的实验与计算结果。对键性质的计算与NLMO分析,采用AMS软件ADF模块完成。计算结果表明,锔的5f轨道对锔-硫键的贡献在高压下显著增强,在11GPa时翻倍。 与[Cm(pydtc)4]–光谱中观察到的变化相比,加压后[Nd(pydtc)4]–的吸收光谱中f-f 跃迁,以及Cm(III)苯六甲酸盐的f–f 跃迁发射光谱的变化较小,这是由于它们的键性质受压力的扰动较小。 这表明,锕系化合物的共价性,即使对同一离子也是复杂的,但研究压力对锕系化合物的影响,可以指导配体的选择。 参考文献: Joseph M. Sperling, Evan J. Warzecha, Cristian Celis-Barros, Dumitru-Claudiu Sergentu, Xiaoyu Wang, Bonnie E. Klamm, Cory J. Windorff, Alyssa N. Gaiser, Frankie D. White, Drake A. Beery, Alexander T. Chemey, Megan A. Whitefoot, Brian N. Long, Kenneth Hanson, Paul Kögerler, Manfred Speldrich, Eva Zurek, […]
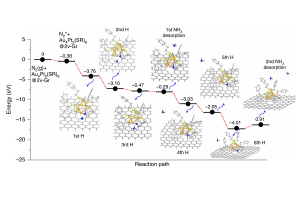
原子精度掺杂控制单团簇催化电化学氮还原(Nat. Comm. 2020)
精确设计亚纳米双金属团簇掺杂,为原子级别调控催化性能提供了机会。而原子级掺杂控制与制备单分散双金属团簇催化剂一直以来都是巨大的挑战。 清华大学李隽教授课题组与新加坡国立大学、美国布鲁克海文国家实验室合作,报道了一种可控的精确掺杂单团簇催化剂合成策略,该催化剂是以缺陷石墨烯为基底,由配体对Au4Pt2团簇进行部分修饰。产生的双金属单团簇催化剂(Au4Pt2/G),具有优异的电化学氮还原活性。理论机理研究表明,N2分子在团簇和石墨烯之间的有限区域被激活。杂原子Pt增强了到N2的LUMO轨道的电子反馈,这在N2活化过程中起着不可或缺的作用。此外,除杂原子Pt外,用Pd代替Pt可以进一步调节单团簇催化剂的催化性能。 为了更直观的了解,移除配体如何改变团簇的电子性质,作者使用AMS软件中ADF模块,计算了Au4Pt2(SR)6和Au4Pt2(SR)8 (R=H)的Kohn-Sham分子轨道能级。去除两个配体,不仅减小了团簇的电子能隙,而且产生了两个分别来自Pt的5d和Au的6s的单占据轨道,而这两个活跃电子可能有助于电子从团簇转移到N2的π*轨道,从而活化N2。 参考文献: Chuanhao Yao, Na Guo, Shibo Xi, Cong-Qiao Xu, Wei Liu, Xiaoxu Zhao, Jing Li, Hanyan Fang, Jie Su, Zhongxin Chen, Huan Yan, Zhizhan Qiu, Pin Lyu, Cheng Chen, Haomin Xu, Xinnan Peng, Xinzhe Li, Bin Liu, Chenliang Su, Stephen J. Pennycook, Cheng-Jun Sun, Jun Li, Chun Zhang, Yonghua Du […]
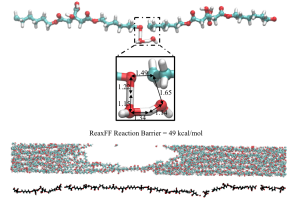
基于加速反应分子动力学的柠檬酸盐聚合物生物降解模拟(J. Phys. Chem. B 2020)
具有弹性的生物可降解聚合物在组织工程领域得到了广泛的关注,而聚酯是用于制造骨组织支架的常用生物材料。最近Adri van Duin等人利用加速反应分子动力学模拟,研究了柠檬酸盐聚合物的反应性。 由于酯和乙醚水解反应的活化能垒较高,反应所需时间相对于计算机模拟时间尺度(一般在ns级别的微观时间尺度)而言非常长,因此往往需要通过特殊的方法,加速分子动力学模拟中化学反应的发生。作者在ReaxFF框架内使用了bond boost加速反应方法(参阅教程),Bond boost参数的选择是通过人为调整得到。乙醚水解比酯水解能垒高,当bond boost参数降低时,乙醚水解几乎停止。另外对聚合物进行了机械拉伸模拟,以确定其拉伸模量,显示出应变相关的行为(另见应力应变教程),发现聚酯比聚醚具有更高的韧性。 己二醇-柠檬酸与两个水分子反应的ReaxFF过渡态结构(顶部),多链聚合物束降解水解,然后施加机械应变(中间),单聚合物链降解(底部)。 参考文献: Nabankur Dasgupta, Dundar E. Yilmaz, and Adri van Duin, Simulations of the Biodegradation of Citrate-Based Polymers for Artificial Scaffolds Using Accelerated Reactive Molecular Dynamics, J. Phys. Chem. B 124, 5311–5322 (2020)
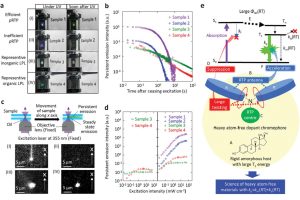
无重元素的高效持久室温磷光分子(Advanced Materials 2020)
持久性(寿命>100 ms)室温磷光(p-RTP)对于最先进的生物成像应用非常重要。发色团与p-RTP相关的物理参数之间关系不明,这导致寻找产率超过50%和寿命超过1s的p-RTP非常困难。日本电气通信大学最近报道了在环境条件下,不含重元素发色团的高效、长寿命p-RTP。由长共轭氨基取代无重原子芳香核,显著加快了磷光发光速率,且与分子内振动的T1态无辐射跃迁无关。设计出的其中一个生色团在环境条件下的RTP产率为50%,寿命为1s。强激发下的余辉亮度至少是传统长余辉发光体的104倍。这表明,实现小规模、低成本、达到衍射极限尺寸的高分辨率门控发射的光电探测器是可能的。 文中通过ADF计算的旋轨耦合强度,详细研究了三重态-单重态间窜跃,以及激发态的辐射跃迁寿命等。 参考文献: Indranil Bhattacharjee, Shuzo Hirata, Highly Efficient Persistent Room‐Temperature Phosphorescence from Heavy Atom‐Free Molecules Triggered by Hidden Long Phosphorescent Antenna, Advanced Materials, 2020, 2001348