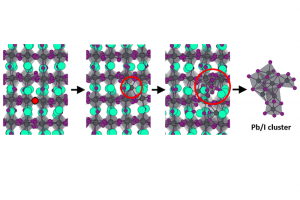
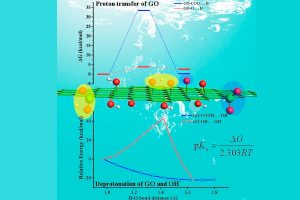
近年来,卤化物钙钛矿引起了人们极大的关注。高转换效率、低生产成本和易于制造,使其成为太阳能电池技术的理想候选。尽管有这些优点,钙钛矿型太阳能电池的商业化,仍受长期稳定性差这个问题所障碍。长期稳定性差是由各种尚未完全了解的材料固有过程造成的。埃因霍温理工大学(TU/E)和宾夕法尼亚州立大学(PSU)的研究人员,在最近的一篇论文中,使用ReaxFF进行反应分子动力学模拟,揭示了卤化物钙钛矿中降解过程的原子细节。 这是首次使用ReaxFF研究卤化物钙钛矿(CsPbI3)的报道。I/Pb/Cs参数由精确的量子力学计算数据作为训练集,使用和蒙特卡罗方法优化获得。所得到的参数集用于研究无机卤化物钙钛矿中的一系列动力学和反应过程。 新的ReaxFF参数能够预测卤化物钙钛矿的适当相和动力学行为。反应分子动力学模拟揭示了钙钛矿晶格分解为PbI2的原子机制。 在碘空位(红点)存在的情况下,铅物种可以离开其在晶格中的位置,形成局部富Pb/I的区域,进一步演化形成Pb/I簇。 参考文献: M. Pols, J. M. Vicent-Luna, I. Filot, A. C. T. van Duin, and S. Tao, Atomistic Insights Into the Degradation of Inorganic Halide Perovskite CsPbI3: A Reactive Force Field Molecular Dynamics Study, J. Phys. Chem. Lett. 12, 5519–5525 (2021).