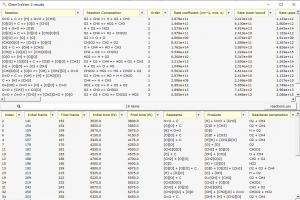
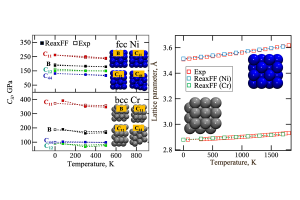
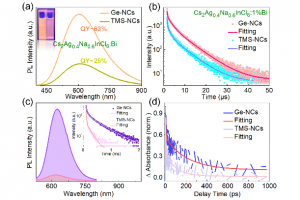
AMS包含全面、完善的计算模拟方法,在原子水平上对分子与团簇、聚合物、低维材料、框架结构材料、多孔材料、宏观流体提供丰富的性质模拟、预测工具。初学者友好的完善图形化操作,可以协助研究者顺利完成建模、计算、作业管理、结果分析、图谱展示。图形化操作界面,支持最新版本的Win、Linux、Mac系统 AMS主要包括分子与团簇第一性原理计算模块ADF,周期结构材料的第一性原理计算模块BAND,反应力场模块ReaxFF,溶解、萃取、蒸馏、共晶等流体热力学模拟模块COSMO-RS(通常需要ADF模块的协助,用于生成分子的coskf文件),工作流与工具集AdvanceWF模块,机器学习势与力场模块ForceField,半经验方法Mopac与DFTB,其中DFTB不仅包含原始dftb.org参数,还包括大量自建参数,覆盖了元素周期表大部分元素。 AMS2022在功能方面的改进 AMS整体算法 AMS提供多种算法驱动,例如分子动力学、巨正则系综蒙特卡洛、Force Bias蒙特卡洛、多尺度模拟、势能面扫描、振动分析等,这些算法,能够灵活调用不同计算模块。 PES Exploration:自动化反应通路搜索算法,使用户能够调用任何计算模块自动探索过渡态和局部能量最小值:过程搜索:找到局域能量最小点,以及它们之间的过渡态搜索初始结构附近的过渡状态Basin hopping寻找局域能量最小值在不同理论水平上,对过渡态和能量最小值点进行精细化计算团簇或表面上的结合位点的确定、可视化案例(使用ReaxFF演示)请点击PES Scan:扫描晶格尺寸、碎片态表面沉积分子动力模拟:用于ALD、CVD、刻蚀模拟,支持气体组分比例定制分子动力学计算均方位移、粘度轨迹回放:对其他轨迹文件,使用指定计算引擎重复其轨迹,并计算相关性质,通常用于比较势能面差异弹性墙壁(纳米反应器):将反应物束缚于纳米球壁内,常在ReaxFF外加电场时使用摩擦学特性(剪切应力):目前只支持命令行模式 ForceField模块 包含Machine Learning Potentials与经典力场GFN-FF、UFF、UFF4MOF、UFF4MOF-II、GAFF、Amber98、Tripos5.2 AdvanceWF模块 ChemTraYzer 2.0:由于ReaxFF自带ChemTraYzer仅适用于小分子化学反应的分析,SCM自行开发了ChemTraYzer 2.0,可用于小分子、大分子、聚合物、非均相等各种类型化学的分析,并能够提供基元反应方程式、反应级数、反应速率常数(采用实验测量单位),并能够对每个反应相关原子演化过程单独显示,以便分析反应轨迹。(详细使用效果请点击)ParAMS:用于训练DFTB参数、ReaxFF力场的图形化工具:将AMS中的计算结果添加到训练集中设定训练集每个样本的权重因子导入样本力场设定需要优化的参数使用CMA-ES优化力场交叉验证生成力场Microkinetics:在从试剂到最终产品的转化过程中,通常涉及许多中间基本反应步骤。这些基本反应步骤具有各自不同的能垒和反应速率常数,它们的整合决定了整个系统的反应行为。通过微观动力学建模,对这样的系统进行研究,可以得到整体反应速率,以及限制整体反应速率的关键因素。包含如下功能:计算温度范围内的反应速率计算不同产物的选择性确定反应的反应级数和表观活化能计算所有反应步骤的速率控制程度处理均相和异相反应应用充分混合或活塞流反应器模拟程序升温脱附模拟同位素的开关ACE Reaction:自动生成反应网络(测试中)ReactMap:确定化学反应中反应物和产物之间的最佳原子映射OLED模拟工具:OLED器件的多尺度建模、气相沉积、计算薄膜中所有分子的电离势、电子亲和力和偶极矩等特性的分布、将数据传输到 Simbeyond 的Bumblebee代码,用于涉及您的材料的 OLED 设备模拟 ADF模块 极化力场:QM/FQ Quantum Mechanics/Fluctuating Charges非弛豫偶极矩激发态激发态间跃迁偶极矩快速激发光谱计算方法POLTDDFT:扩大到绝大多数元素仅特征值自洽 GW (evGW)、G3W2配体场密度泛函(LFDFT)新功能:ESR g-张量双峰,XMCDMP2考虑自旋轨道耦合 COSMO-RS模块 改进了对多物种流体热力学的处理,例如不同的质子化和解离状态、聚集(使用溶剂)、构象异构体COSMO-RS-PDHS参数:增加了计算带电体系所需的长程静电项(如下图所示) BAND模块 -D4色散修正泛函虚晶近似:允许某些原子位以一定比例掺杂费米面的图形化显示自旋轨道耦合DOS电子能量密度函数周期结构材料的结合能XPS APPLE&P 极化力场模拟,可用于带电体系,如电解质(例如电池中的电荷迁移率)、离子液体类体系的分子动力学模拟,案例参考:离子体系的分子动力学模拟 Zacros & Zacros-post模块 动力学蒙特卡洛模拟 Quantum ESPRESSO模块 更新到7.0版 免费试用 http://www.scm.com/free-trial