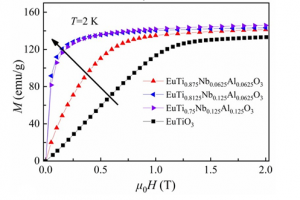
概述 低温大磁熵变材料在太空科学领域和氢气液化的应用中具有重要的应用价值,因此高效低温磁制冷材料及其相关问题研究,也就成为当前国际上凝聚态物理领域的热点课题之一。稀土Eu基钙钛矿氧化物EuTiO3因具有大磁矩和低磁相变温度,是一类性能优良的低温磁制冷材料。但是,EuTiO3基态是反铁磁,如果能够选择的合金元素掺杂改变其磁基态,则可在低驱动场下实现大磁熵效应。中科院理化所的沈俊教授课题组和南京大学王敦辉教授、陆海鸣副教授课题组合作,结合第一性原理和实验研究了在Ti位同时掺杂Nb和Al对EuTiO3体系电子结构和低场磁热性能的影响。该研究成果为优选高性能低温稀土磁制冷材料提供了新思路。 图1 三种不同Nb和Al掺杂量的Eu(Ti,Nb,Al)O3的总态密度和分波态密度图 研究亮点 通过第一性原理计算,作者发现Nb和Al的共掺使得晶格常数增加且反铁磁的EuTiO3转变为铁磁态。从图1所示的Eu(Ti,Nb,Al)O3体系的总态密度(TDOS)和分波态密度(PDOS)图可以看出,Nb和Al的掺杂引起的自旋极化导致自旋向上和自旋向下不完全对称,巡游的Ti的3d和Nb的4d能级横跨费米能级,发生了绝缘体—金属转变。从PDOS图可以看到,巡游的Ti的3d电子、Nb的4d电子和Al的3p电子都和局域的Eu的4f电子发生交互作用,产生RKKY作用。Nb和Al的掺杂引发的反铁磁-铁磁相变可用四阶扰动理论(fourth-order perturbation theory)来解释。磁相互作用由两个独立的部分组成:RKKY作用和Anderson超交换(前者具有金属特性而后者具有半导体特性)。EuTiO3的态密度图中费米能级处的态密度为0,没有RKKY作用,此时,通过无磁的Ti4+离子的Anderson超交换在EuTiO3中占主导地位,体系为反铁磁。当Nb和Al置换掺杂进来后,自旋极化和交换分裂出现并增加,同时费米能级也移向高能量处,使得Anderson超交换耦合减弱而RKKY作用增强并占主导地位,因此体系呈铁磁态。 图2 不同成分化合物2K时的M(H)曲线以及与其他化合物等温熵变和有效制冷能力的比较 作者发现EuTiO3的磁化强度在磁场低于1 T时线性增加并在2 T时饱和(左图),而三种不同成分的Eu(Ti,Nb,Al)O3在1 T时均达到饱和且饱和磁化强度均高于EuTiO3,这说明Nb和Al的掺杂不仅可以显著降低临界场还可以提高其低场磁化强度。右图是1 T磁场下不同体系等温熵变和有效制冷能力的比较。三种不同成分的Eu(Ti,Nb,Al)O3的等温熵变约为-15.1 J/kg·K,比EuTiO3高了近40%。此外,EuTi0.8125Nb0.125Al0.0625O3和EuTi0.75Nb0.125Al0.125O3的有效制冷能力分别达到了88.1和86.0 J/kg,不仅比EuTiO3高了近315%,也都比其他化合物体系的有效制冷能力高。 总结 本文利用AMS软件的BAND模块,进行了通过不同价态阳离子共掺杂改变EuTiO3电子结构和磁基态的研究。研究发现,Nb和Al共掺杂不仅诱发了EuTiO3的绝缘体—金属转变,还引发了反铁磁-铁磁相变。实验结果还表明,共掺杂样品在1T低磁场下的磁熵变都达到了15 J/kg·K,是EuTiO3的1.4倍;特别是EuTi0.8125Nb0.125Al0.0625O3的有效制冷能力达到了88 J/kg,是EuTiO3的3倍。该工作明确了元素掺杂的EuTiO3体系中磁交换作用机理,为液氦温区磁制冷提供了有竞争力的候选材料。 参考文献 Huicai Xie, Wenxia Su, Haiming Lu, Zhaojun Mo, Dunhui Wang, Hao Sun, Lu Tian, Xinqiang Gao, Zhenxing Li, Jun Shen, Enhanced low-field magnetocaloric effect in Nb and Al co-substituted EuTiO3 compounds, J. Mater. Sci. Technol., 118, 128, […]