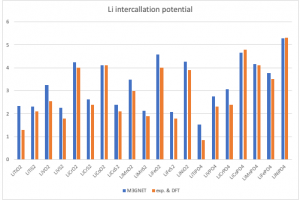
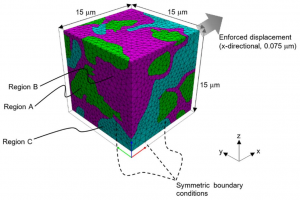
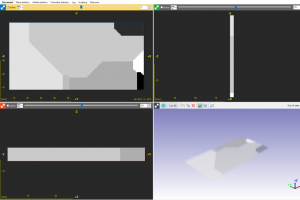
Simpleware 软件中包含许多有助于分析纤维数据的工具,主要用于理解取向分布和计算各种统计数据。本教程将展示无需单独进行图像分割的纤维分析核心功能。 数据来源:Simpleware 软件数据文件夹 FibreOrientation 1. 背景图像中心线 1.1 准备数据 打开 Fibres.sip 项目文件,3D 视图自动生成体积渲染模型 图1:Simpleware ScanIP 中生成的纤维材料体积渲染模型 1.2 由背景图像生成中心线 打开 Measurements — Centrelines — Create centrelines 工具,确认 Input 是 Active background (Fibres),设置捕捉纤维中心的阈值范围为 128-255。(对于中空纤维,可能需要一些前处理,例如使用 Laplacian of Gaussian 滤波器。)通过计算穿过单个纤维的体素乘以体素间距设置接近于纤维直径的 Feature diameter (mm) 参数。针对本例数据,该值设为 0.03。勾选 Isolated lines (fibres),显示 Pruning sensitivity、Joining angle threshold 和 Remove short lines 的设置选项,保持默认即可。依次点击 Create 和 OK。放大 3D […]