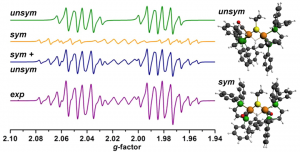
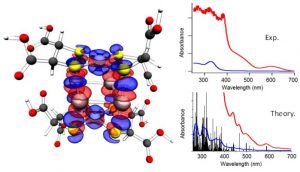
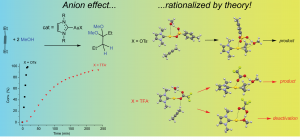
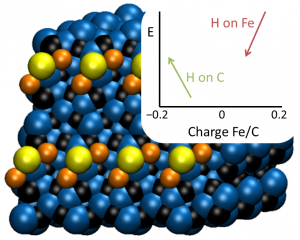
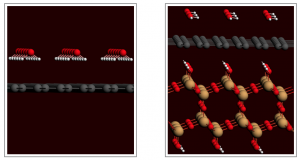
参考文献: W. Wang, M. J. Nilges, T. B. Rauchfuss, and M. Stein, Isolation of a Mixed Valence Diiron Hydride: Evidence for a Spectator Hydride in Hydrogen Evolution Catalysis J. Am. Chem. Soc. 135, 3633-3639 (2013) 由于进一步发展氢经济的需求,双铁化合物,仿[FeFe]-H2已经作为更廉价、更具可持续性的HER催化剂被广泛研究。University of Illinois and the MPI for Dynamics of Complex Technical Systems的研究者分离出并表征了混合价态双铁氢。为仿生HER催化领域展开新的视角。 本文使用ADF的ZORA-DFT计算得到的几何结构、IR位移、EPR参数与实验结果非常相符,从而洞悉对不对称和对称异构体的自旋分布。 关键词:EPR, IR, ZORA, spin distribution