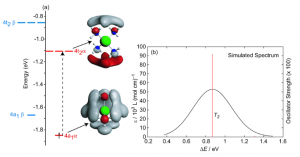
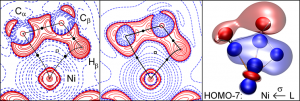
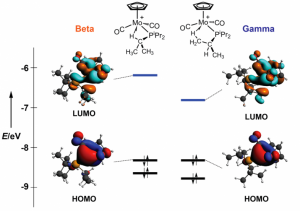
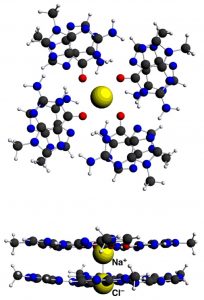
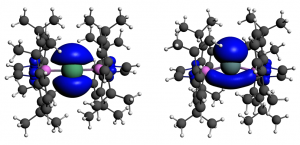
参考文献: E. Zurek, P. P. Edwards and R. Hoffmann, A Molecular Perspective on Lithium-Ammonia Solutions. Angewandte Chemie International Edition, 48(44), 8198 (2009). 美国康奈尔大学Roald Hoffmann教授是诺贝尔化学奖得主,是ADF的忠实用户。本文介绍了关于锂-氨溶液的详细计算分析工作。锂金属-氨溶液从200年前首次发现的时候,就一直被人们好奇于它的“fine blue colour”。 最终结论是:这些体系不是“金属铵”,而是碱阳离子以及溶液中电离的电子,这增加了这个体系的神秘性。电子态的怪异性、卓越的颜色性质,都能够使用TDDFT进行研究。 锂离子的氨溶液结构优化(使用COSMO溶剂化模型),以及电子吸收光谱的计算,都使用ADF完成。其中一些模拟中,涉及到自由基体系,溶剂化电子分布在氨分子周围。开壳层电子吸收谱的计算也还不算普遍。光谱和涉及的电子转移的轨道的可视化是在ADF-GUI中完成的。 使用ADF功能: excited states (with TDDFT), un-paired electrons (open-shelled systems)