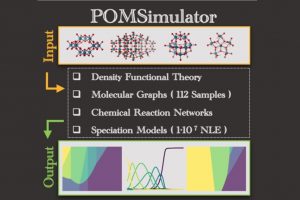
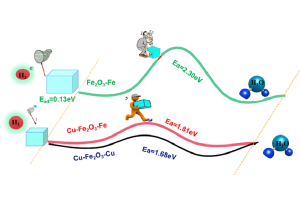
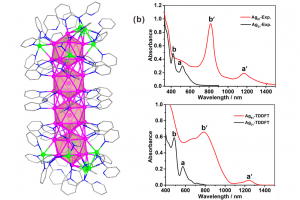
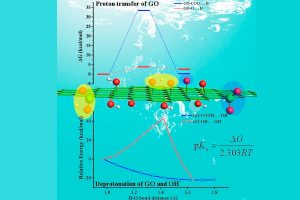
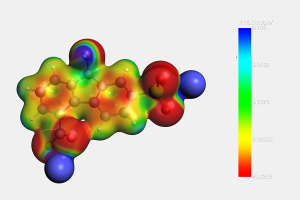
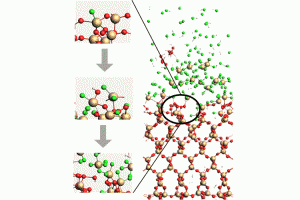
由于描述所有化学平衡的高度复杂性,多酸在水溶液中的自组装问题,仍然是一个需要研究的课题。实验技术在这一问题的研究上取得了巨大的成功,但还需要更多的努力来促进这些金属氧化物在材料科学中的应用(详见综述)。去年,ICIQ的研究人员开发了一种名为POMSimulator的模拟器,用于描述小钼团簇的水相形态。 为了充分再现钼和钨氧化物的水相形态,今年代码得到进一步扩展,对122种金属氧化物结构进行了优化,并用ADF计算了它们的解析频率。结果被传递给POMSimulator,该模拟器预测了所考虑的所有多金属氧酸盐的平衡常数。理论常数与实验常数呈线性关系,表明ADF的自由能可以直接重新标度。 参考文献: [1] Petrus, E.; Segado, M.; Bo, C. Nucleation Mechanisms and Speciation of Metal Oxide Clusters, Chemical Science 2020, 11, 8448 [2] Petrus, E.; Bo, C. Unlocking Phase Diagrams for Molybdenum and Tungsten Nanoclusters and Prediction of Their Formation Constants, J. Phys. Chem. A 2021, 125, 5212