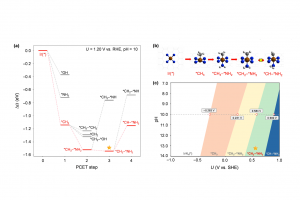
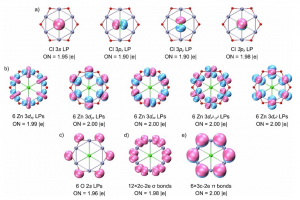
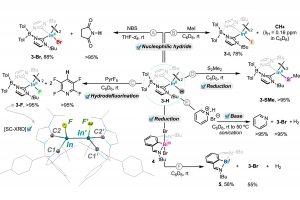
研究背景 甲烷(CH4)作为天然气的主要成分,是极其丰富的碳基资源。然而,由于甲烷具有极其牢固的 C-H 键(键能高)以及非极性特征,其在温和条件下的选择性活化与高值化利用一直是化学与催化领域极具挑战性的科学难题。传统的甲烷转化工艺往往依赖于高温高压的蒸汽重整或甲醇中介胺化,不仅能量消耗巨大,且伴随着极低的碳利用效率与显著的温室气体排放。近年来,电化学甲烷活化技术重塑了这一领域,实验研究已证实可在温和条件下实现甲烷的氨化反应生成甲胺,但微观反应机理尚不明确,且传统氧化物催化剂的活性位点结构复杂,深层反应机制与选择性调控规律仍不清晰。石墨烯负载的氮掺杂碳单原子催化剂(M-N-C)凭借其明确且可调控的活性中心,成为阐明反应机理的理想平台。其中,具备 IrN4 活性中心的 Ir-N-C 单原子催化剂因 5d 过渡金属 Ir 具备空间延伸且能量灵活的 d 轨道,且对氧亲和力相对较弱,在选择性活化甲烷及抑制析氧副反应(OER)方面展现出独特潜力。基于此,清华大学化学系肖海教授团队在期刊《Nano Research》上发表了题为“Potential-induced bis-axial coordination in Ir-N-C single-atom catalyst delivers selective C-N coupling for efficient methane amination”的最新研究论文。作者采用巨正则系综密度泛函理论(GCE-DFT)结合周期性能量分解分析(PEDA/EDA-NOCV)及活化应变模型(ASM),系统揭示了阳极电势下 IrN4 单原子活性位点的轴向配位动态演变机制及其对 C-N 偶联选择性的调控本质。 研究内容 作者利用恒定电位下的巨正则系综密度泛函理论(grand-canonical ensemble density functional theory, GCE-DFT)计算,系统解析了工况条件下 IrN4 活性中心随电势和 pH 变化的结构演化规律(图1)。在 U = 1.20 V v.s. RHE 和 pH = 10 的典型阳极环境中,IrN4 位点展现出独特的化学选择性:CH4 优先吸附活化脱氢生成单轴配位的 *CH3 […]