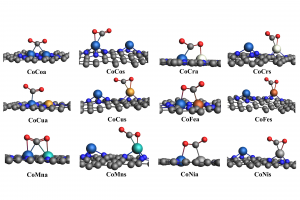
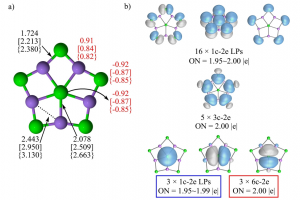
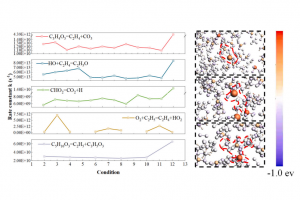
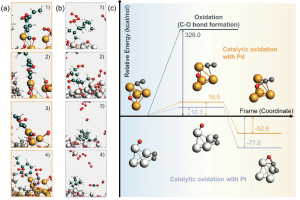
研究背景 电催化 CO2 还原反应(CO2RR)能够在较温和条件下将 CO2 转化为高附加值化学品,是当前碳资源转化与能源化学领域的重要研究方向。其中,乙烯、乙醇等 C2 产物不仅附加值更高,也更能体现催化剂在 C–C 耦合步骤上的调控能力。相比 C1 产物,C2 产物的生成涉及更多中间体和更复杂的反应分叉,因此,如何从原子尺度揭示催化剂结构、反应路径与产物选择性之间的关系,一直是 CO2RR 理论研究中的关键问题。 双原子催化剂由于具有相邻双活性中心和可调电子结构,在促进 CO2 活化、关键中间体稳定以及 C–C 耦合方面展现出独特优势,近年来受到广泛关注。围绕这一方向,相关研究构建了 Co 基双原子氮掺杂石墨烯模型,系统比较不同双原子组合和不同位点构型下的稳定性、反应活性与产物选择性,为高选择性 CO2RR 催化剂设计提供理论依据。 在这类研究中,计算平台的适配性同样十分重要。AMS 软件中的 BAND 模块能够直接处理二维周期体系,适用于石墨烯、氮掺杂碳材料及表面催化模型;同时,结构优化、吸附能计算、溶剂模型、自由能分析以及电荷和轨道相互作用分析可以在同一平台内完成,因此非常适合开展这类涉及构型、吸附、路径、机理等全流程的理论研究。 研究内容 云南大学刘世熙课题组采用 AMS 计算软件中的 BAND 模块,设计了两种构型的 DACs 首先计算了不同构型载体的形成能以及其对金属原子的吸附能,结果表明a构型载体具有更低的形成能,同时对过渡金属原子的吸附能力也更强。此外,通过 Bader 电荷分析、PDOS 及 COOP 计算,进一步验证催化剂中 M1 和 M2 的 3d 轨道与 N 的 2p 轨道之间存在明显的成键作用,从而揭示了催化剂的稳定性。 图1. CoN3-M2N3@NGr-a(a)以及CoN3-M2N3@NGr-s(b)的俯视图;CoN3-M2N3@NGr-a(c)以及CoN3-M2N3@NGr-s(d)的侧视图 采用 BAND […]