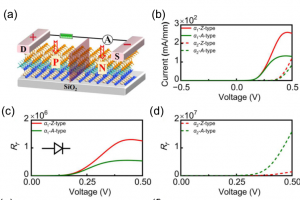
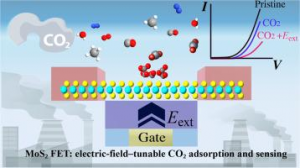
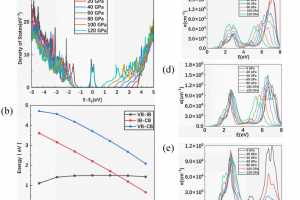
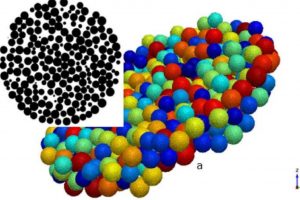
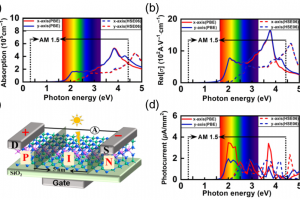
研究背景 二维 MA2Z4 材料因具有优异的热稳定性、力学稳定性、可调电子结构和较高载流子迁移率,已成为低维电子学和光电子学中的重要候选体系。Janus 结构通过在材料上下表面引入不同原子层,打破面外对称性,为调控能带、极化、输运和光响应提供了新的自由度。此前 Janus MoSi2N2P2 的稳定性和光学性质已有理论预测,但其电子输运、场效应调控和光电响应等器件行为仍需进一步阐明。 研究内容 该研究首先构建了 α1 和 α2 相的 MoSi2N2P2、Mo(SiNP)2-A 和 Mo(SiNP)2-B 单层,并对其进行结构优化。声子谱、AIMD 热稳定性和弹性常数计算表明,这些二维单层具有良好的结构稳定性、热稳定性和力学稳定性。其中 MoSi2N2P2 单层兼具较好的柔性和尺寸稳定性,适合进一步用于柔性、超薄纳米电子器件的设计。 图 1. α1 和 α2 相的 MoSi2N2P2、Mo(SiNP)2-A 和 Mo(SiNP)2-B 单层的俯视图和侧视图(不同颜色小球分别表示 Mo、Si、N 和 P 原子) 在电子结构方面,能带和态密度结果显示,MoSi2N2P2 单层具有较窄带隙,有利于低电压下载流子注入和调制。进一步计算载流子迁移率,发现 Janus MoSi2N2P2 在 x 方向的迁移率高于 y 方向;其中 α1 相在 x 方向的载流子迁移率相较 MoSi2N4 有明显提升,并可与 MoSi2P4 的性能相媲美。 图 2. MoSi2N4、MoSi2P4 以及不同 Janus MoSi2N2P2 相关单层的投影能带结构和态密度。 随后,构建基于 Janus MoSi2N2P2 的 p-n 结二极管,并分别考察 zigzag 与 armchair 方向的电子输运特性。结果表明,该类器件具有明显整流效应;其中 α1 相 Z-type p-n 结在中等掺杂浓度下峰值电流密度达到 270.37 mA/mm2,并表现出负微分电阻现象,显示其在低维整流器和多值逻辑器件中的应用潜力。 […]