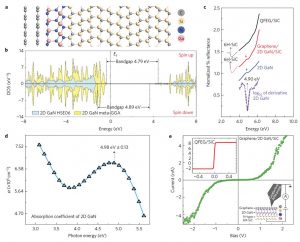
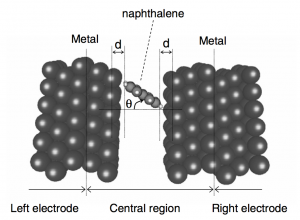
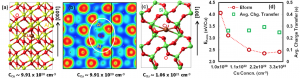
摘要 文章(Surface Science 600 (2006) 5080–5083)研究了单个π共轭分子吸附或夹在两个电极上的电流特性,尤其是载流子通过有机/金属界面的注入。首次使用第一性原理方法研究了分子的取向和电极材料对电流对影响:过去的电流计算都是假设分子与电极之间通过共价键连接。研究模拟的两个体系中,萘分子夹在金或铝电极之间。 首先,通过分子的电流与分子的取向有关系,这表示电子主要通过π通道(也就是分子的π轨道与电极电子轨道的交叠)传输;其次,金-萘-金体系的电流比铝-萘-铝体系的电流强,这表明金比铝更适合做电极。 参考文献 First-principles study on current through a single p conjugate molecule for analysis of carrier injection through an organic/metal interface Kenji Toyoda *, Kiyoshi Morimoto, Kiyoyuki Morita Advanced Technology Research Laboratories, Matsushita Electric Industrial Co., Ltd, 3-4 Hikaridai, Seika-cho, Soraku-gun, Kyoto 619-0237, Japan Surface Science 600 […]
![FIG. 1. (Color online) Structures of the geometry optimized 1-nm NWs. (a)–(c) show the unterminated [100], [110], and [111] NWs, (d)–(f) show the respective O-terminated NWs. Cu is orange and O is red.](https://www.fermitech.com.cn/wp-content/uploads/2016/11/atk_for_famous_corp001_01-300x227.png)