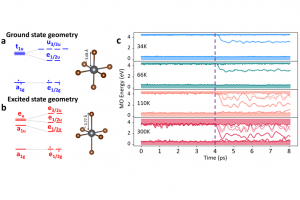
Cs4PbBr6属于0D金属卤化物家族,具有分离和独立的[PbBr6]4-八面体。在不同的报道中,其纳米晶粒与块体形式,在室温下不发光或发绿色光,对绿色发光的起源或猝灭的机制没有明确一致的结论。要解开这个谜,意大利理工学院(IIT)纳米化学系的研究人员研究了构成该材料的孤立卤化铅八面体部分的电子结构,然后在不同温度下对Cs4PbBr6固态材料的2x2x2超胞进行了从头算分子动力学研究。使用AMS软件ADF模块在DFT(TDDFT)/PBE/DZP理论水平上,对单个八面体性质进行研究,评估了基态和最低激发态几何结构之间的差异,以及自旋轨道耦合(SOC)对能级和电子跃迁的影响,发现激发态的几何弛豫主要是八面体内Pb-Br键的轴向伸长,SOC贡献导致最低激发态的三重态和单重态特征混合,从而导致光学上允许的自旋禁止跃迁,与光致发光的激发和发射光谱一致。 对完整Cs4PbBr6的分子动力学模拟表明,电子与Pb-Br伸展运动相关的声子发生耦合,导致光致发光的猝灭,因此光致发光仅出现在低温下,这与实验数据是一致的。CsBr空位缺陷被认为是绿色发射的主要原因之一,它的加入再次导致了发光的猝灭,因此相互连接的八面体是唯一可能产生绿色发射的缺陷。综上所述,在纯材料中,由于热淬火室温发射被抑制,观察到的绿色发射可能只是导致PbBr6八面体之间互连的结构缺陷的结果,而不是空位或其他点缺陷。 参考文献: U. Petralanda, G. Biffi, S. C. Boehme, D. Baranov, R. Krahne, L. Manna*, and I. Infante, Fast Intrinsic Emission Quenching in Cs4PbBr6 Nanocrystals, Nano Lett. 21, 8619–8626 (2021)