
机器学习力场:模拟真实体系
机器学习力场(ML-FFs)可以进行接近从头算精度的模拟,但是模拟体系的尺寸和动态模拟时间尺度则大大超过了从头算,更接近真实情况。使用 QuantumATK 中的机器学习力场功能可以生成新颖的晶体和非晶材料、合金、界面和多层堆叠的真实的复杂结构,模拟热性能和机械性能、扩散和表面过程。用户可以使用预先训练的机器学习力场库,或使用自动化、高效的训练和仿真工作流程开发新的机器学习力场。

QuantumATK中的计算引擎
QuantumATK 计算模拟引擎能够在一个平台上使用多种模拟方法进行原子级建模和模拟[1]:先进的密度泛函理论(DFT)(平面波基组的 DFT-PlaneWave 或 原子轨道基组的 DFT-LCAO )、半经验量子力学方法、传统力场(内置 300 余力场数据库)和机器学习力场。所有模拟引擎在材料特性、动力学、过渡态搜索(NEB)、几何结构优化和其他模拟等方面均使用一个计算框架,这为 QuantumATK 上训练和使用机器学习力场提供了绝佳的集成环境。
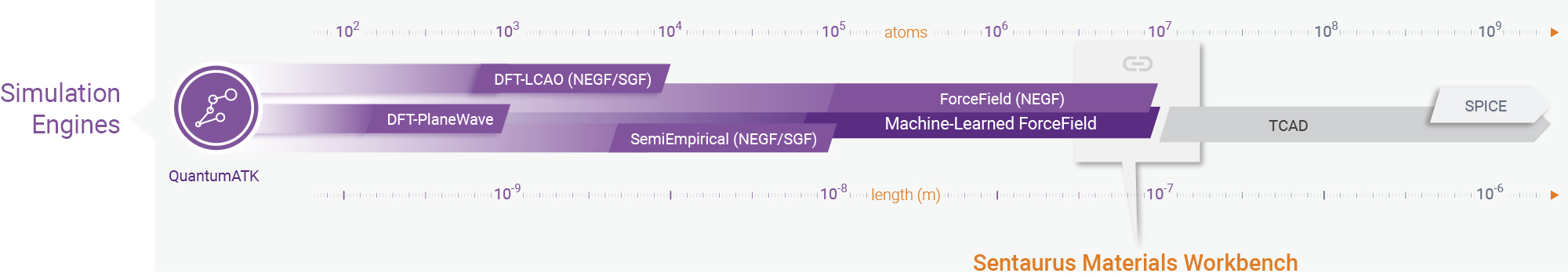
使用机器学习力场对大型真实体系进行动力学模拟
- 机器学习力场比 DFT 快 1000 到 10000 倍,因此能够对包含超过 100000 个原子的真实、新颖、复杂的体系进行动力学模拟(通常 DFT 可以达到的 100 原子体系);
- 机器学习力场为多元素材料、界面等异质体系和远离平衡的体系(包括非晶态材料、相变或化学反应)提供了几乎和从头算一样的精度;
- 使用 QuantumATK 提供的机器学习力场的自动化计算流程,可以比传统力场更容易完成动力学计算。一般来说,复杂体系的传统力场计算过程往往很复杂,需要反复测试和调整计算过程与参数。

机器学习力场的应用实例
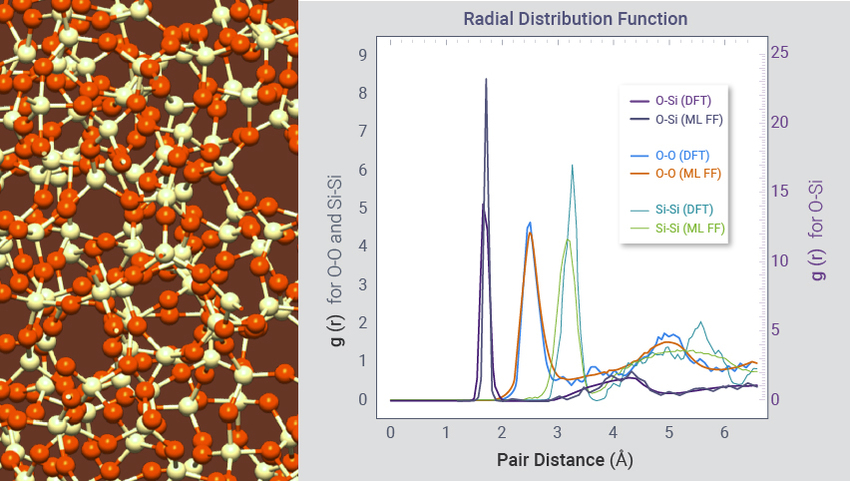
生成非晶材料结构
为 PCRAM、ReRAM 和 FeRAM 等新型存储器、太阳能电池和其他应用领域生成非晶结构。在这个例子中,80 ps-ML-FF-MD 在 11 分钟内生成了 600个原子的 am-SiO2 结构,而在 16 个核上用 DFT-MD 生成 72 个原子的结构需要 10 天。用 ML-FFs 获得的结构参数与 DFT 和实验结果吻合良好。
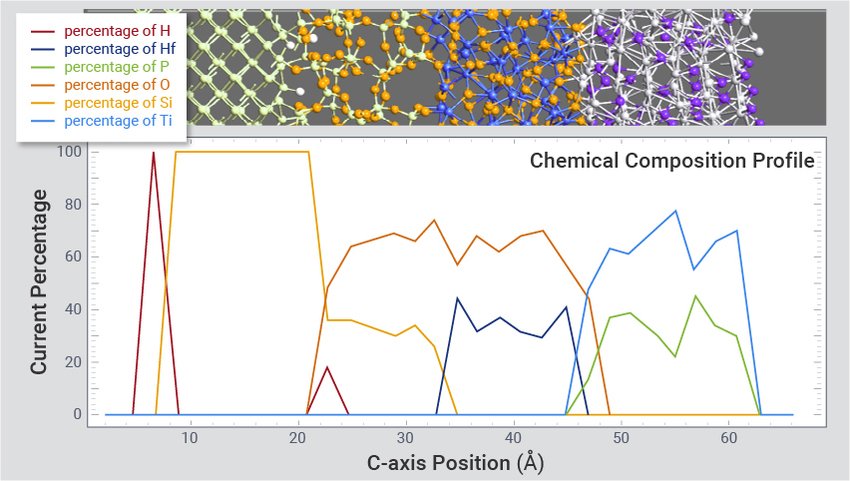
界面和多层堆叠的模拟
为半导体研发构建和优化复杂的晶体和非晶界面以及多层堆叠结构,例如高 k 金属栅(HKMG)(使用图形界面的多层结构建模工具构建)和 MRAM 磁性隧道结工程。该示例显示了几乎无缺陷的 c-Si|am-SiO2|am-HfO2|am-Ti2N HKMG堆叠的结构。
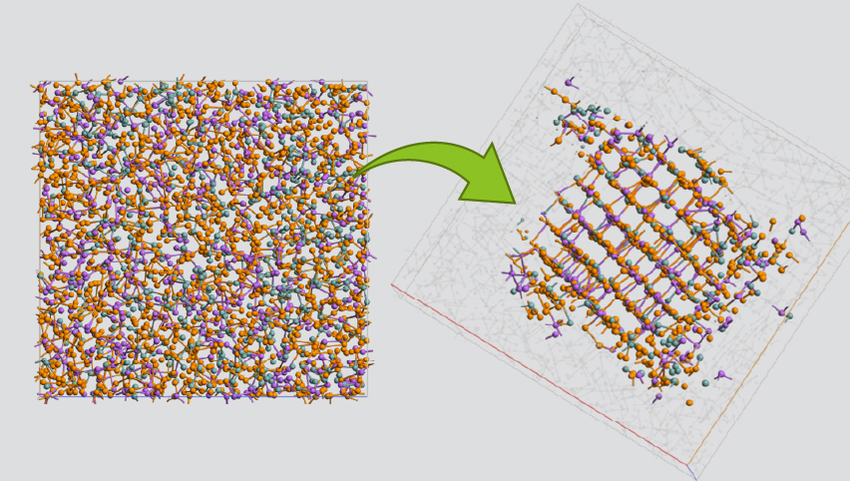
玻璃态非晶材料的模拟
产生用于光电应用的含有杂质的玻璃态非晶材料。在本例中,使用 ML FF-MD 模拟了在 2500 K 下具有 Na 杂质 (Na2O)2(SiO2)40000 的 120000 原子的大型玻璃模型。

结晶和非晶化过程
使用 ML FF-MD 研究大规模体系中的纳秒时长的结晶和非晶化过程,例如 PCRAM 新型存储器应用。本实施例描述了2520个原子相变合金材料Ge2Sb2Te5 的结晶。
热性能模拟
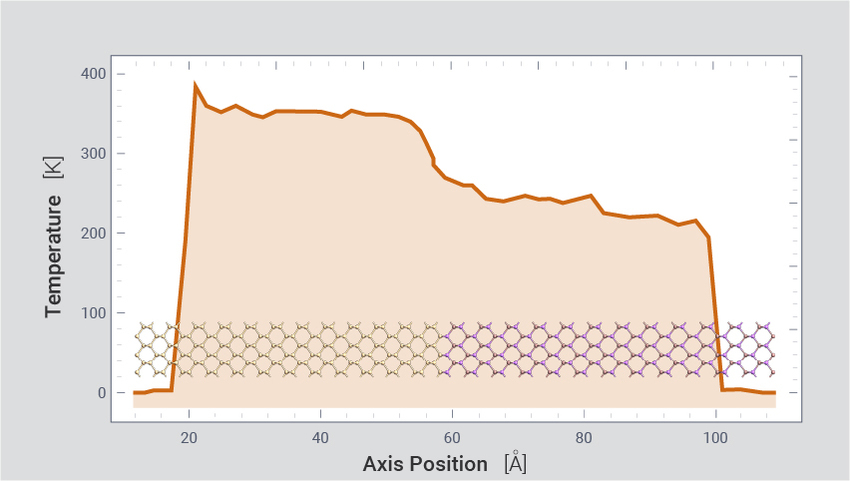
使用 ML FFs 进行非平衡分子动力学(RNEMD)来模拟导热,研究诸如 PCRAM 器件中的自发热和散热。示例包括模拟块体 Ge2Sb2Te5(2300 个原子)、Ge2Sb2Te5|Si(882个原子)和 Si|GaAs(864个原子)界面、单层 MoS2(108000 个原子)中的导热。计算值与实验和已知的 DFT 结果一致。
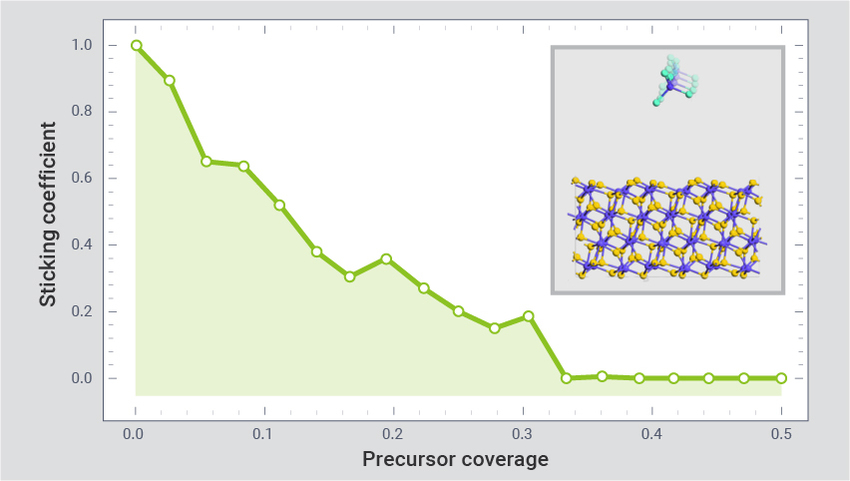
表面过程建模
使用经过专门训练的 ML FFs 进行 MD 模拟,研究 ALD 和 ALE 过程。此示例显示了 ALD 过程的模拟:HfCl4 在 HfO2 上的沉积 面积为 4.5 nm2 的表面。前驱体分子吸附能与 DFT 结果一致。获得的粘滞系数和覆盖值可以用作特征尺度模型的参数,以优化 ALD 的产率。
内置现成的机器学习力场库

QuantumATK 提供了 Moment Tensor Potential(MTP),与其他机器学习力场相比,该力场具有较高而且强壮的精度和较低的计算成本[2,3]。用户可以使用预先训练好的高质量 MTP 库(参见材料列表[4,5]),也可以通过使用下文所述的自动生成工作流程开发新的 MTP,用于新材料、界面和表面工艺。
自动且高效的生成机器学习力场
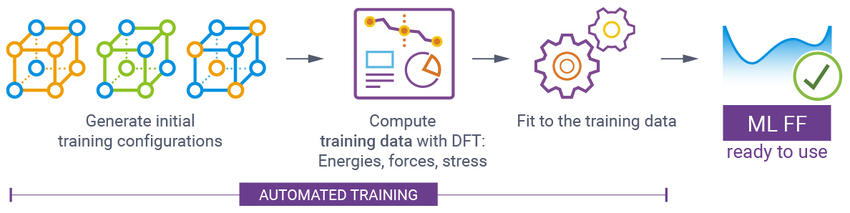
基本工作流程
- 自动生成训练结构(基于晶体结构的随机位移)
- 使用 DFT 计算训练数据,如能量、力和应力
- 执行机器学习,拟合训练数据
- 验证生成的 ML FFs 并优化超参数
- 使用 ML FFs 进行实际模拟,如 MD 或 fbMC 的动力学模拟、NEB 过渡态搜索和几何结构优化
- 计算可以使用 OpenMP、大规模 MPI 并行或混合并行
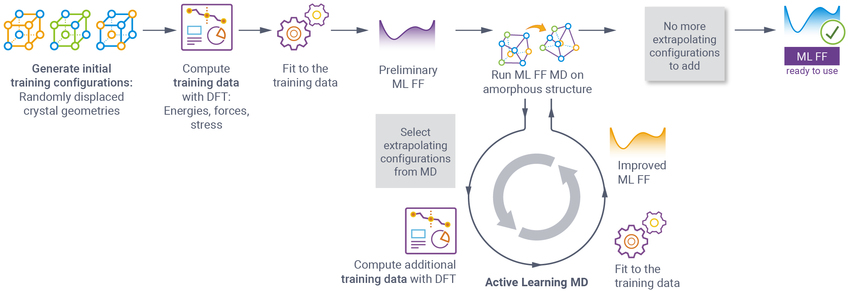
高级主动学习工作流程
- 通过在 MD 模拟过程中主动添加训练结构和 DFT 训练数据,改进使用基本工作流程生成的初始 ML FFs
- 自动迭代过程基于结构外推的特定阈值
- 推荐用于
- 非晶态体系
- 材料界面
- 高温体系
- 表面过程建模
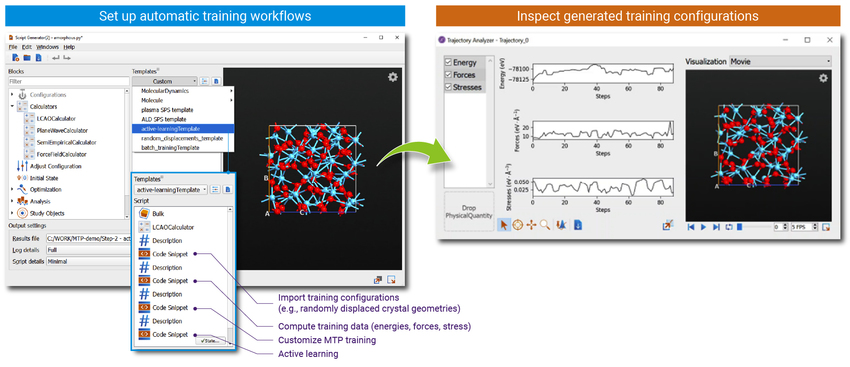
图形用户界面与流程模板
- 使用自动训练工具和 GUI 模板[6]进行以下体系的操作:
- 晶体和非晶块体材料
- 材料界面
- 分子
- 使用轨迹分析工具 GUI 检查自动生成的训练结构
- 通过将计算值与以下可用的实验和 DFT 数据进行比较,验证生成的 ML FFs:
- 径向/角度分布函数( RDF 和 ADF)
- 弹性常数
- 中子散射因子
- 化学组分概貌
- X 射线散射
QuantumATK优势
- 自动生成用户友好的训练数据,为特定应用量身定制
- 确保所需的训练数据和时间最少
- 在大多数情况下,不需要计算成本很高的从头算 MD
- 为复杂体系提供高质量、精确的 ML FFs
- 不同计算引擎都使用统一接口
- 训练所用的 DFT 可以在 LCAO 和平面波基组之间轻松切换
- 将 ML FFs 与传统力场、DFT 或半经验计算方法相结合,以进一步提高特定应用中的精度
常见问题
机器学习力场的价值
- 为什么不能使用 DFT 运行所有的模拟?
- ML FFs 比 DFT 快 1000 到 10000 倍,但仍然可以达到几乎同样的精度。ML FFs 的动力学模拟将模拟时间从几天减少到几分钟,从几年减少到几小时,从而能够对 DFT 不可能模拟的包含 1000-100000 个原子和更多原子的真实、新颖和复杂体系进行动力学模拟。
- 为什么不使用高效的传统力场运行所有需要的模拟?
- 传统力场仅可以用于特定的材料体系,或者可以将它们结合起来描述不同材料之间的界面,不过应用非常有限且不准确。
- 传统力场通常对远离平衡体系不太精确,这是描述非晶材料、相变或化学反应常遇到的问题。
- 我需要使用机器学习吗?与拟合一个新的传统力场相比呢?
- 传统力场适用于相对简单的材料,为复杂材料拟合精确的传统力场非常困难,事实上,对于多元素材料或界面(特别是金属和半导体之间)等异质体系几乎不可能。
- 可以用 ML FFs 模拟的最大原子数是多少?
- QuantumATK 团队已经测试了对 100000 个原子体系进行纳秒时长的分子动力学模拟。根据可用的计算资源,大型体系也可以用 ML FFs 进行模拟。
QuantumATK中的机器学习力场
- QuantumATK 是如何实现 MTP 机器学习力场的?
- QuantumATK 团队自己实现了 MTP,并不是使用文献[2,3] 中的原始 MTP 程序包。团队预计 QuantumATK 中的 MTP 与原始程序的效率类似,但没有进行明确的效率比较,因为它们还依赖于 MD 模拟引擎。
- 使用 QuantumATK 生成机器学习力场需要多长时间?
- 如果有 2-4 个集群节点(32-72 个核心),以下是获得几种体系的高质量 ML FFs 的时间:
- 晶体结构(1-3 种元素)需要 1-2 天(依赖于材料的复杂性)
- 界面和非晶材料需要 1-2 周(包括主动学习 MD,依赖于复杂程度)
- 更复杂的体系需要数周(例如 3 个以上的元素、各种界面或 ALD 等特殊应用)
- 在所有情况下,最耗时的部分是生成训练结构和使用 DFT 计算训练数据。对训练数据的拟合可以在几个小时内或最多一天内完成。生成的训练结构可以在为类似材料(例如元素相同但化学计量比不同的体系)开发ML FFs时重复使用。
- 如果有 2-4 个集群节点(32-72 个核心),以下是获得几种体系的高质量 ML FFs 的时间:
- 预先训练或生成的机器学习力场可以在哪些软件上运行 MD 仿真?
- 生成的 ML FFs 仅用于在 QuantumATK 中进行 MD 模拟。QuantumATK 提供高性能、大规模并行、最先进的 MD 模拟方法。
- 与其他软件解决方案相比,QuantumATK 在机器学习力场方面有哪些优势?
- QuantumATK 提供了一种用户友好、自动和高效的训练数据生成方法,而不是手动生成
- 这对于高质量的 ML FFs 和减少训练时间和数据量至关重要;通常 DFT 参考数据量可以减少 10-100 倍
- 独特的自动化 QuantumATK 流程专为特定应用定制,如分子表面沉积或界面
- 其他方案中的 ML FF 模型(NNP、GAP等)比 QuantumATK[2,3] 中使用的 MTP 模型精度更低、速度更慢
- QuantumATK 表明,机器学习力场对于复杂材料(如非晶态合金、涉及多种材料的界面以及分子表面沉积)也具有很高的准确性和效率,而不仅仅是简单材料
- 为什么生成训练结构过程可以不用从头算MD模拟?
- 使用从头算 MD 生成训练配置是一个计算成本非常高的过程。根据 QuantumATK 团队的经验,这通常会导致比实际需要多得多的 DFT 计算,而得到的 MD 结果系列结构通常非常相关和相似。QuantumATK 主要使用自己的训练方法验证了这一点,该方法使用晶体结构的原子随机位移和应变来产生结构。此方法可以很好的描述固体性质,如晶格常数、声子和中等温度下的弹性常数,而无需添加从头算 MD 模拟产生的结构。然而,对于无定形或其他更复杂的结构,包含从头算 MD 训练数据更好。
参考
- S. Smidstrup, T. Markussen, P. Vancraeyveld, J. Wellendorf, J. Schneider, T. Gunst, B. Vershichel, D. Stradi, P. A. Khomyakov, U. G. Vej-Hansen, M.-E. Lee, S. T. Chill, F. Rasmussen, G. Penazzi, F. Corsetti, A. Ojanpera, K. Jensen, M. L. N. Palsgaard, U. Martinez, A. Blom, M. Brandbyge, and K. Stokbro, “QuantumATK: An integrated platform of electronic and atomic-scale modelling tools”, J. Phys.: Condens. Matter 32, 015901 (2020). arXiv: 1905.02794v2.
- A. V. Shapeev, “Moment tensor potentials: a class of systematically improvable interatomic potentials”, Multi-scale Model. & Simul. 14, 1153 (2016).
- Y. Zuo, C. Chen, X. Li, Z. Deng, Y. Chen, J. Behler, G. Csányi, A. V. Shapeev, A. P. Thompson, M. A. Wood, and S. Ping Ong, “Performance and cost assessment of machine learning interatomic potentials”, J. Phys. Chem. A 124, 731 (2020).
- 机器学习力场特性: https://www.synopsys.com/silicon/quantumatk/resources/feature-list.html#MLforcefield
- QuantumATK包含的训练好可以直接使用的力场: https://docs.quantumatk.com/manual/ForceField.html#pretrained-moment-tensor-potential-mtp-parameter-sets
- 训练机器学习力场自动化工具和GUI图形用户界面使用教程: https://docs.quantumatk.com/tutorials/mtp_hfo2/mtp_hfo2.html