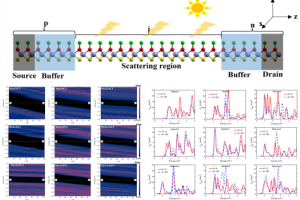
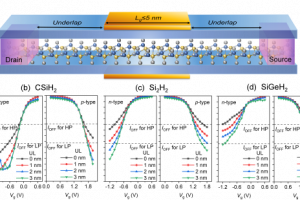
研究背景 随着集成电路器件尺寸持续向亚 5 nm 尺度推进,传统硅基 MOSFET 在短沟道效应、量子隧穿及功耗控制等方面面临严重瓶颈,难以满足未来高性能与低功耗芯片的发展需求。如何在极限尺度下实现高驱动电流、低泄漏及良好的栅控能力,并能够与现有硅基工艺相兼容,有效利用现有器件制造技术,成为下一代晶体管研究和应用亟须解决的关键问题。 研究内容 该研究针对超短沟道 MOSFET 性能提升这一核心问题,基于一类具有硅基工艺高度兼容性的二维单层材料 MNH2(M, N = C, Si, Ge),在保证高载流子输运能力的同时实现与现有工艺体系的良好兼容,为器件实际应用提供基础。该工作系统地开展了器件物理与量子输运研究,并构建适用于亚 5 nm 尺度的双栅 MOSFET 模型,深入分析其输运特性与性能极限。 图 1. MNH2单层几何结构及电子性质 研究结果表明,通过引入 Underlap 结构对沟道势垒进行调控可有效抑制短沟道效应并降低漏电流,从而在保证高导通电流的同时实现低功耗运行。通过系统分析不同结构参数对器件性能的影响,揭示了超短沟道条件下电流输运由隧穿机制向热发射机制转变的关键物理过程,从机理上实现器件性能的优化与提升。 图 2. 二维 MNH2 MOSFET 器件结构及输运特性 在器件性能方面,基于 MNH2 材料构建的 MOSFET 在亚 5 nm 尺度下表现出显著优势。在 5 nm 栅长条件下,器件导通电流最高可达 4157 μA/μm(n 型)和 3527 μA/μm(p 型),显著优于多数已报道的二维半导体器件。同时,器件的亚阈值摆幅最低可达 58 mV/dec,接近传统 MOSFET 理论极限,表明其具有优异的栅控能力和开关特性。 图 3. 二维 MNH2 MOSFET 开态电流特性比较 在动态性能方面,该类器件的延迟时间和功耗延迟积均远低于国际半导体技术路线图(ITRS)2028 指标要求,体现出良好的高速与低功耗特性。此外,在栅长进一步缩小至 3 nm 时,器件仍能够保持较高性能并满足高性能应用需求,显示出优异的尺度可扩展能力。与现有二维材料相比,该体系在驱动电流、功耗及综合性能方面具有明显优势,同时具备良好的工艺兼容性,为实际应用提供了重要基础。 值得注意的是,该体系呈现出良好的 n-p 对称性,适于用作 CMOS 器件。该研究系统证明了 MNH2 二维材料在亚 5 nm MOSFET 中的应用潜力,实现了高性能、低功耗与工艺兼容性的综合平衡,为后硅时代晶体管设计提供了新的技术路径和理论依据。 图 4. 二维 MNH2 MOSFET 功耗延迟积特性比较 总结 该研究面向先进微电子与集成电路领域,在下一代超大规模集成电路、低功耗高性能芯片以及新型二维半导体器件中具有重要应用前景。由于 MNH2 材料体系在电学性能与硅基工艺兼容性之间实现了良好平衡,可作为亚 5 nm 及以下节点 MOSFET 的潜在候选沟道材料,为先进制程技术提供新的解决方案。 在高性能计算、人工智能芯片以及移动终端等领域对器件功耗与性能的要求不断提升,该研究提出的材料与器件有望显著提升芯片能效比。同时,由于其 n 型和 p 型器件性能均衡,具备良好的 CMOS 集成潜力,可支持未来新型逻辑器件的发展。随着后硅时代电子技术的持续演进,该成果有望在纳米电子器件及高端芯片制造中发挥重要作用,具有良好的应用前景与推广价值。 参考文献 Hao-Ran Hu, Yan-Dong Guo, et al. Monolayer MNH2 (M, N = C, Si, Ge): Enabling sub-5-nm MOSFETs compatible with silicon-based manufacturing. Physical Review Applied, 2026, 25, 014058 […]