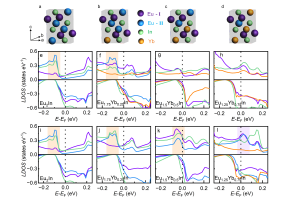
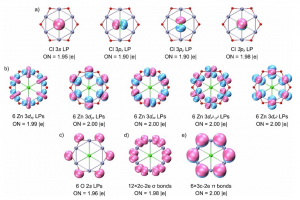
研究背景 相变是凝聚态物理与功能材料领域的核心研究方向,相变性质直接影响材料的应用性能。在新兴的磁制冷技术中,一阶相变伴随潜热、熵变数值大,但普遍存在磁/热滞损耗;二阶相变无滞后、循环稳定性优异,制冷温区更宽,但磁熵变峰值偏低。近年来,科研人员发现在磁制冷材料中引入外来元素,不仅能调节相变温度,在某些情况下甚至还能改变材料本身的相变性质。例如,本征的 Eu2In 铁磁材料发生一阶相变,但当掺杂非磁性 Yb 形成 Eu1.25Yb0.75In 时,相变转变为二阶相变;当两个本身都发生一阶相变的铁磁材料 Pr2In 和 Nd2In 形成 PrNdIn 合金时,竟出现所谓的“边界一阶相变”。针对掺杂改变相变性质的科学问题,现有研究仅聚焦单一母相的本征电子结构,相变性质演化的微观电子起源缺失,从而制约了相变性质的定向设计。南京大学陆海鸣副教授课题组以稀土 RE2In 合金为研究对象,结合第一性原理计算与朗道-金兹堡自由能展开理论,系统探究了相变性质演化的电子起源。以 Yb 掺杂的 Eu2In 体系为模型体系,定量引入模-模耦合系数b作为相变判据(b为负对应一阶相变,b为正则对应二阶相变),通过计算费米能级附近的态密度,揭示驱动相变性质演化的特定位点轨道杂化作用;并借助化学掺杂、静水压双重调控手段验证机制,澄清 PrNdIn “边界一阶相变”的本质为弱一阶相变;最终基于杂化工程理论设计出新型一阶磁制冷材料 ErPrIn,实现了从机理阐释到材料预测的闭环。 研究内容 图 1、Eu2-xYbxIn的无磁态电子结构与模-模耦合系数。(a)Eu2In、(b)Eu1.75Yb0.25In、(c)Eu1.50Yb0.50In、(d)Eu1.25Yb0.75In在费米能级附近的总态密度、和(e)模-模耦合系数随Yb掺杂浓度的变化关系 Eu2In 金属间化合物因兼具巨磁热效应、低热磁滞后等优势,成为理想的磁制冷材料。其具有正交 Co2Si 型结构,存在两种不等价的 Eu 位点——Eu-I 和 Eu-II。由于 Yb 掺杂的 Eu2In 体系有充足的相变性质演变的实验数据可供参考,作者选取 Eu2-xYbxIn(x 为 Yb 掺杂浓度)作为模型体系。通过第一性原理计算,发现当Yb 逐渐替代 Eu 时,Yb 优先占据与 In 键合更强的 Eu-II 位点。通过计算不同 Yb 浓度下 Eu2-xYbxIn 的无磁态电子结构与模-模耦合系数 b(图 […]