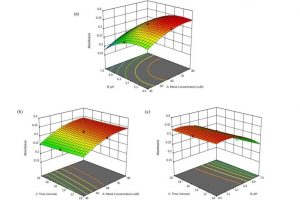
使用有机化合物的化学传感器,可以作为识别水环境中金属离子的优秀可选方案。传感器的优化过程严重影响所设计传感器的性能,在本研究中,采用响应面法(RSM)成功开发了一种利用有机化合物(即氨基硫脲连接乙酰吡嗪,TLA)识别不同环境水样中Co2+离子的高灵敏度和选择性比色传感器系统。作者建立的模型得到了成功的优化,并且具有统计显著的独立的变量(p<0.05),在pH值为5.3、100:70µM TLA/Co2+浓度、二甲基亚砜/水反应体积比8:2、反应时间15 min,达到最佳识别。在最佳条件下,TLA传感器可识别浓度低至1.637µM的Co2+离子,这低于火焰原子吸收光谱法(FAAS)的检测限。理论方法支持实验数据,并对化学传感系统中TLA-Co2+的机械非共价相互作用进行了表征和预测。最后,本研究中产生的所有积极结果表明,TLA可作为识别水中Co2+污染的替代和可比探针,具有成本效益、可移动且易于处理、无需特殊培训且环保。 在理论方面,利用COSMO-RS方法对TLA在其介质中的分子极化或相互作用进行了理论研究;通过计算Fukui函数来确定何种原子更易位于亲核区和亲电区;采用TDDFT方法验证了所建议的配合物结构以及TLA与Co2+离子之间的相互作用;结合NCI-RDG方法,TD-DFT方法提出的模型进一步用于描述该化学传感系统中TLA和Co2+离子之间发生的相互作用类型。 参考文献: Hakimah Ismail, Mohammad Norazmi Ahmad & Erna Normaya, A highly sensitive and selective thiosemicarbazone chemosensor for detection of Co2+ in aqueous environments using RSM and TD/DFT approaches, Scientific Reports volume 11, Article number: 20963 (2021)