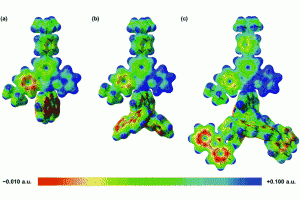
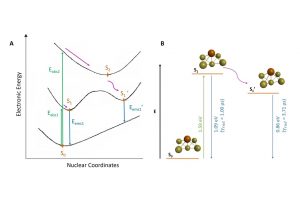
香港大学化学系分子功能材料研究所,最近首次设计并合成了一类新的含C^C^N配体的咔唑金(III)树状大分子,其固态薄膜的光致发光量子产率高达82%,辐射衰减速率常数高达105 s−1。通过变温发射光谱、时间分辨光致发光衰减和计算研究,发现这些金(III)树状大分子表现出热激活延迟荧光(TADF)性质。并基于这些金(III)树状大分子,制备出溶液加工型有机电致发光二极管(OLED),其最大电流效率为52.6cd A−1,最大外部量子效率为15.8%,高功率效率为41.3 lm W−1。并记录了这些OLED的运行稳定性,基于零代和第二代树状大分子的器件在100 cd m-2下的最大半衰期分别为1305小时、322小时。 为了更深入地了解这些包含三齿配体的金(III)树状大分子的电子结构,以及吸收和发射的起源,作者进行了密度泛函理论(DFT)和TDDFT 计算。对基态、激发态分子结构、激发态,以及参与激发的分子轨道进行了系统性的分析,其中480-520 nm 的激发主要由HOMO → LUMO贡献。HOMO是主要位于咔唑部分的π轨道,而LUMO是主要位于中央苯环和C^C^N配体的吡啶基部分的π*轨道,因此,HOMO → LUMO跃迁可以认定为 LLCT [π(咔唑) → π*(C^C^N)] 跃迁,三种结构的吸收带及其光谱分配与实验结果趋势一致。 为了更深入地了解增加咔唑基单元对环金属化配体电子密度的影响,还计算了三种结构的基态静电势面。显然,由于树状咔唑取代基的吸电子作用,较高代咔唑基树枝的引入,导致更缺电子的吡啶基,这降低了辅助咔唑基N-供体配体上的电子密度,减少了其给电子的能力。咔唑基N-供体配体的供体强度较差,会使金属中心更加缺电子,通过σ效应从C^C^N钳形配体中吸取更多的电子密度。电子富集较少的金属中心对金属 dπ 轨道起到稳定作用,这反过来会导致 π* (C^C^N) 轨道较小程度的不稳定。总体而言,第二种结构的 HOMO-LUMO能隙 (3.04 eV)比第一种 (3.06 eV) 和第三种 (3.09 eV) 的能隙略窄,与实验趋势非常吻合。 为了更深入地了解发射态的性质,使用非限制性PBE0对T1进行了几何结构优化。三种分子的自旋密度主要位于 C^C^N 配体的咔唑基部分、中心苯环和吡啶基部分,从而支持发光态的LLCT [π(咔唑) → π*(C ^C^N)]特性。发射波长采用S0和T1优化几何结构之后的能量差近似,发射波长从第三种分子 (507 nm) 到第一种分子 (530 nm)与第二种分子 (534 nm)显现出红移特点,这与实验中观察到的趋势一致。 为了进一步了解 TADF 过程中涉及的激发态,使用包含Tamm-Dancoff 近似 (TDA) 的 TDDFT 优化了三种分子的S1和T1的几何结构。计算得到三种分子ΔE ST值分别为 0.005、0.006 和 0.004 eV。为了进一步了解第一种分子的的 […]