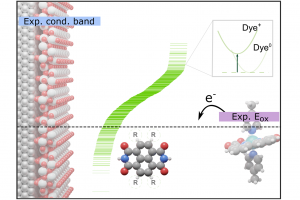
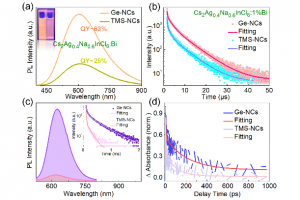
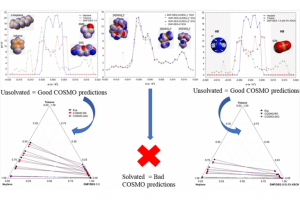
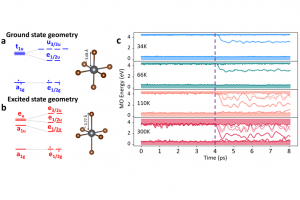
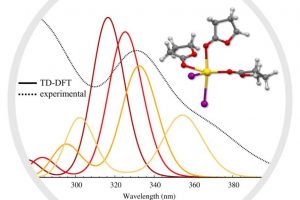
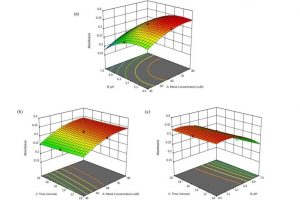
染料敏化光电化学电池(DS-PEC)是一种很有前途的可持续燃料生产系统,这种设计的巨大优势是系统的模块化。特别值得一提的是,可以通过轻微的结构调整来调整染料的光学和电化学性质,如基态氧化电位(GSOP)。在最近的一篇论文中,有人提出了一种快速、自动化的工作流程,用于自动筛选数千个候选分子,以识别有前途的染料。 为了设计该工作流程,对许多最先进的电子结构方法进行了评估,以及通过氧化反应的吉布斯自由能(产物和反应物的溶液相吉布斯自由能之差)或溶剂化染料的垂直电离能计算GSOP的不同方法。 使用DFT计算溶液相吉布斯自由能,包括使用COSMO或COSMO-RS方法考虑溶剂效应。此外,还评估了在Kohn Sham和GW水平下计算的垂直电离电位对GSOP的近似。对于苝类染料,考虑氧化后的几何弛豫和电子溶剂效应非常重要,而其他热效应可以忽略。结合精度和计算效率,优化工作流包括使用SQM方法(GFN1 xTB)执行的几何优化、包括COSMO的单点DFT计算和COSMO-RS热力学计算。 参考文献: J. Belić, A. Förster, J. P. Menzel, F. Buda, and L. Visscher, Automated assessment of redox potentials for dyes in dye-sensitized photoelectrochemical cells, Phys. Chem. Chem. Phys. 24, 197-210 (2022)