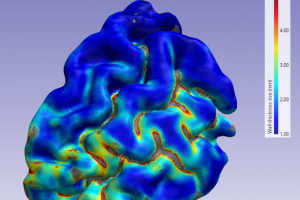
在 Simpleware ScanIP 中可以使用壁厚工具对获得的掩膜或面模型进行厚度统计和 3D 可视化厚度分布。本教程将向您展示如何利用壁厚工具研究大脑皮层预分割区域的神经解剖学皮层厚度,该指标被认为与认知能力相关,可用于阿尔茨海默病等脑部疾病的诊断。 本教程所用数据文件路径 C:\Program Files\Synopsys\Simpleware\U-2022.12\Data\WallThickness 1. 生成表面渲染 点击File — Open 打开 CerebralCortex.sip 项目文件。 壁厚工具是使用 3D 视图中对象的三角曲面渲染计算和展示厚度分布。 确认 Dataset browser 下的 Grey matter 掩膜为可见状态。点击 3D preview — Model Setup — Setup model,打开 Model configuration 对话框。在 General 标签,选择 Binarise before smoothing 模式,勾选 Use smart mask smoothing,其他保持默认。在 Surface settings 标签,勾选 Use triangle smoothing for masks,设置 […]