
1,使用FDE(Frozen-Density Embedding)方法计算粒子迁移与能量传递
主讲人:Michele Pavanello, Rutgers University
时 间:2015年2月24日 北京时间23:30
人 数:暂无限制
参考文献:
[1] P. Ramos and M. Pavanello, Quantifying Environmental Effects on the Decay of Hole Transfer Couplings in Biosystems J. Chem. Theory Comput., 10, 2546-2556 (2014).[2] A. Solovyeva, M. Pavanello, and J. Neugebauer Describing long-range charge-separation processes with subsystem density-functional theory J. Chem. Phys., 140, 164103 (2014).
[3] M. Pavanello, T. van Voorhis, L. Visscher, and J. Neugebauer, An accurate and linear-scaling method for calculating charge-transfer excitation energies and diabatic couplings, J. Chem. Phys. 138, 054101 (2013).
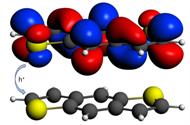
DFT对于某些问题,例如上图所示体系,容易出现问题。在上图所示体系,分子轨道一般是局域的,但DFT计算却非常容易产生非局域的HOMO、LUMO,同时分布在两个分子区域上——这并不符合化学直观,实际上也是不正确的。这个问题一般容易被忽略。FDE很好地在DFT的框架下,解决了这个问题。
Michele是ADF中FDE方法的开发者。本次在线课程涉及复杂体系的电荷、激发能传递动力学。该方法将电荷、激子、自旋局域化到体系的特定子区域。这些绝热区域之间的耦合决定了电荷的迁移、能量的传递、电荷的分离,以及其他与有机电子学和光伏相关过程。Michele将介绍FDE的理论以及操作实例。
报名方式:
http://www.scm.com/Doc/Teaching/ADFWebinars.html
在线课程录像: