2015年5月13日,国家超级计算深圳中心与费米科技将举办为期1天的“ADF软件高级培训暨计算化学与计算材料学研讨会”。将为大家提供一个计算化学领域面对面讨论与交流的机会。 培训班邀请国际著名计算化学专家Fedor Goumans博士为研究者深入讲解ADF软件在科研中的运用技巧,同时邀请国内从事计算化学研究的专家分享详解计算化学领域最新进展。 同期的培训班将为大家详细讲解并演示ADF软件的基本操作以及高级技巧。培训期间国家超级计算深圳中心会提供临时应用账号,用于登陆超算中心主机。请自带笔记本电脑,体验ADF在超级计算中心的高速运行。 主讲教师:Fedor Goumans博士 ADF开发组 高级技术支持 荷兰自由大学计算化学博士、伦敦大学学院博士后,在计算化学领域有丰富的科研经验,同时有很强的实验学科背景。 日期:2015年5月13日(星期三) 地点:国家超级计算深圳中心(深圳市南山区西丽大学城学苑大道1068号 西侧) 费用:免费(食宿交通等费用自理) 日程表: 在线报名: 国家超级计算(深圳中心) 费米科技(北京)有限公司 2015年4月

ADF Highlight:龙虾煮熟为啥变红?(2005,JACS)
参考文献: A. A. C. van Wijk,. A. Spaans, N. Uzunbajakava, C. Otto, H. J. M. de Groot, J. Lugtenburg and F. Buda, Spectroscopy and Quantum Chemical Modeling Reveal a Predominant Contribution of Excitonic Interactions to the Bathochromic Shift in α-Crustacyanin, the Blue Carotenoprotein in the Carapace of the Lobster Homarus gammarus. Journal of the American […]
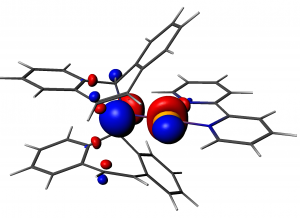
ADF Highlight:稳定的缩醛胺金属配合物 (2005,Science)
参考文献: Torsten Büttner, J. Geier, G. Frison, J. Harmer, C. Calle, A. Schweiger, H. Schönberg and H. Grützmacher, A Stable Aminyl Radical Metal Complex. Science, 307 (5707), 235 (2005). Grützmacher教授带领的ETH Zurich研究组,分离并表征了一种能够支撑稳定的N中心自由基的铑缩醛胺自由基配合物。文中使用ADF的相对论ZORA方法,Spin-unrestricted GGA计算超精细及核四极耦合。Grützmacher教授对ADF的评述:”The ADF calculations by our theoretician Carlos Calle played a crucial role in the EPR assignments”。 点评:对于过渡金属配合物,相对论效应往往比较重要,ZORA方法是ADF的特色功能。对于EPR的计算,采用Slater基函数能够提高精度。
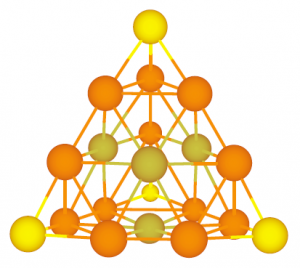
ADF Highlight:锕系-惰性气体配合物、Au20 (2003,Science)
参考文献: J. Li, B. E. Bursten, B. Liang and L. Andrews, Noble GasÐctinide Compounds: Complexation of the CUO Molecule by Ar, Kr, and Xe Atoms in Noble Gas Matrices. Science 295, 2242 (2002). J. Li, X. Li, H.-J. Zhai and L.-S. Wang Au20: A Tetrahedral Cluster. Science 299, 864 (2003). 西北太平洋国家实验室李隽博士(现任清华大学教授)及其合作者,使用ADF的相对论密度泛函研究了锕系-惰性气体络合物 CUO(Ng)x (Science, 2002, 295, […]
ADF搜索过渡态典型案例
ADF搜索过渡态典型案例 以F2与CH4的反应作为案例来阐释ADF搜索过渡态的计算过程(整个过程的文件下载点击此处) ADF搜索过渡态一般分为如下几个步骤: 优化反应物结构; 猜测反应路径(使用Linear Transit功能,以后简称为LT,让反应物通过某一个或两个参数的线性变化,到达产物); 计算Linear Transit得到的最高点(即近似的过渡态“鞍点”)的频率; 使用Transition State Search(以后简称为TSS)功能搜索真正的精确的过渡态; 计算TSS得到的过渡态的频率,校验是不是有且仅有一个虚频,并且该虚频对应的振动模式分别向反应物、产物方向振动; 从过渡态的虚频沿着正负方向振动一帧(或三帧)分别得到倾向反应物和产物的结构,分别优化之,分别得到反应物和产物的稳定结构,并与一开始设想的反应物、产物结构对比,从而确定是单步反应还是多步反应。

ADF Webinar(费米科技免费在线培训 第2期):ReaxFF模拟金属催化碳纳米管生长
主讲人: 刘俊 ADF技术支持 日 期:2015年3月26日(周四) 14:00-15:30 人 数: 25人
ADF Webinar(SCM免费在线培训 2015年2月)
1,使用FDE(Frozen-Density Embedding)方法计算粒子迁移与能量传递 主讲人:Michele Pavanello, Rutgers University 时 间:2015年2月24日 北京时间23:30 人 数:暂无限制
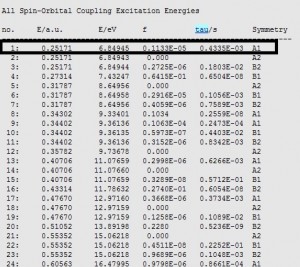
计算荧光、磷光的寿命
1,激发态的基本计算,参考“文献重现:锌酞菁的基态与激发态计算(第二部分)” 2,用户可以根据自己的需要(或文献的建议),修改泛函、基组。 3,对于计算寿命,更改为如下的特殊设置,即可得到相应的寿命数据: Main菜单的设置:Relativity (ZORA) 设为:Scalar Properties——Type of Excitations设为:Spin-Orbit (Pertubative) 在output文件中,可以搜索“tau”,即得到寿命数据(例如下图所示): 在各个不可约表示中,可以分别找到这些激发态对应的信息:例如是单重态或三重态等信息: 对于磷光或荧光,可以分别找到对应的数据。 备注: 荧光和磷光的吸收峰、发射峰的计算,参考“文献重现:锌酞菁的基态与激发态计算(第二部分)”即可,而在这些计算中,如果要同时计算寿命,参数则应做如上的改动。 Relativity (ZORA) 设为:Scalar,表示考虑旋轨耦合效应。考虑该效应之后,自旋不再是守恒量,而只是近似守恒,也即是说,不再有严格意义的单重态或三重态,而只是近似为单重态或三重态,分别对应荧光和磷光。因此根据用户自己关心的发光类型,去找到对应的近似单重态和近似三重态即可。 第一图中的All Spin-Orbital Coupling Excitation Energies列表为考虑 Spin-Orbital Coupling微扰之后的激发能,第二图中的数据在out文件中,出现在第一图数据之前,为考虑Spin-Orbital Coupling微扰之前的数据——在考虑Spin-Orbital Coupling微扰之前,自旋是守恒量,电子态是严格的单重态或三重态。 更多案例参见费米科技WIKI-ADF
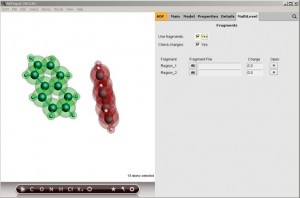
如何计算粒子迁移率
粒子迁移率对于有机电子器件例如场致发射晶体管(OFET)、有机发光二极管、光伏电池非常关键。载流子从一个位置迁移到另一个位置,迁移率主要由转移积分决定。ADF可以直接计算转移积分。作为简化的例子,本例计算两个萘分子之间的电子迁移率。
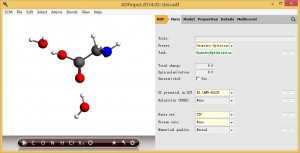
如何计算氨基酸在水溶液中原子的电荷分配
说明:氨基酸在水中通常会形成H键。因此不能使用通常的溶剂化模型如COSMO、SCRF等等来描述,而应该将溶剂分子直接地与溶质分子同等地考虑在内。但溶剂分子的热运动导致溶剂分子相对于溶质的位置比较难以确定。 精确的处理这类问题,需要进行第一性原理分子动力学或者第一性原理的蒙特卡洛模拟,得到溶剂分子相对于溶质分子的位置。这样工作量比较大。 一个比较节省的,可靠性也较高的方式,是将溶剂分子按可能形成氢键的位置预先摆放在溶质分子附近,通过几何结构优化,找到溶剂分子相对于溶质分子的位置。然后基于这样的几何结构计算原子的电荷分配。 我们以甘氨酸举例。