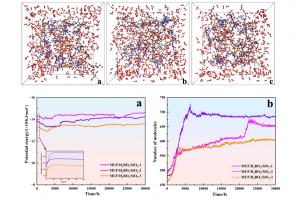
研究背景 可发性聚苯乙烯(EPS) 泡沫因其优异的隔热性、稳定性和低成本等特性,在建筑保温领域占据主导地位。然而,其高度易燃性导致严重的火灾隐患,燃烧时产生的有毒烟雾更是火灾致死的主要原因。阻燃 EPS 泡沫板能有效抑制火灾初期的火势蔓延,显著降低材料燃烧时的热释放速率和有毒烟气产生。因此,提高 EPS 的阻燃和抑烟性能至关重要。 研究内容 本研究由沈阳理工大学和辽宁工程技术大学等单位合作,基于机器学习势方法,使用 AMS 软件中的 ML Potential 模块来完成燃烧模拟工作,其中的 M3GNet 经过 SCM 公司训练优化,成为普适性较好的一种势,默认情况下即可模拟 89 种元素的体系,支持用户针对特定体系进一步优化训练。 作者构建三聚氰胺改性脲醛树脂、纳米二氧化硅、硼酸阻燃涂层晶胞模型,通过模拟手段从微观层面阐明了体系在燃烧过程中通过气相及凝聚相的协同阻燃机制,为高性能 EPS 阻燃涂层的设计提供了理论依据与实践指导。 图 1 涂层分子模型 本研究采用NVT系综模拟阻燃材料的燃烧过程。总模拟步数为120,000步,时间步长为0.25 fs。 温度控制采用Nosé-Hoover(NHC)热浴方法,阻尼常数为10 K/fs,具体设定为:298 K(10,000步)→ 2500 K(40,000步)→ 3500 K(60,000步)→ 4000 K(10,000步)→ 298 K。 在模拟过程中,0-2500 fs 阶段涂层体系内的势能在增加,说明该阶段体系内发生吸热反应;在 2500-7500 fs 阶段体系内的势能在缓慢降低;在 7500-30000 fs 阶段体系的势能趋于平衡。在 7500 fs 内随着温度的升高,三种涂层体系内的总分子数量在快速增加,7500-30000 fs 内,体系内分子的数量上下波动减弱,说明体系内燃烧逐渐减弱,分子的消耗和生成逐渐趋于平衡。 图 2 […]