本研究介绍了一种基于 MRI 创建 PF 关节软骨 3D 模型的方法,帮助更好地了解滑车发育不良、相关病理和 PF 关节一致性等信息以辅助诊断和治疗 PF 患者,特别是复发性髌骨脱位患者。
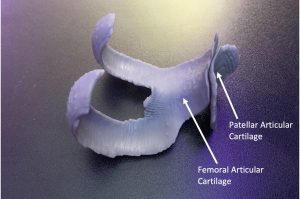

本研究介绍了一种基于 MRI 创建 PF 关节软骨 3D 模型的方法,帮助更好地了解滑车发育不良、相关病理和 PF 关节一致性等信息以辅助诊断和治疗 PF 患者,特别是复发性髌骨脱位患者。
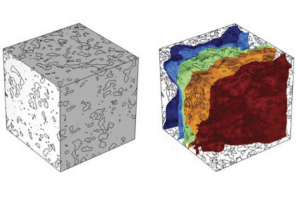
概述 耐腐蚀性能对于钢筋混凝土结构的耐久性至关重要。特别地,抗氯离子侵蚀性能主要与保护混凝土的电阻率(ER)有关。计算电阻率时要考虑的电导池常数由理论获得,在大多数情况下为 2πa,但应根据测试条件进行修改。本研究基于 X 射线Micro CT图像创建三维孔隙模型,模拟多孔水泥基体中连通孔隙内离子传导引起的电流流动,并将数值模拟评估与传统 Wenner 法测量结果进行比较。 实验 圆柱形混凝土试样直径 100 mm,高 200 mm,水灰比为 0.65。使用比重为 3.16 g/cm3 的普通硅酸盐水泥,在 20°C 的室温下养护 28 天。对尺寸为 2 mm 的硬化水泥浆体小试样进行 X 射线 CT 扫描。 表1:混凝土拌合物的配合比 在测试试样前,将其浸入水箱并在真空条件下完全饱和。采用 Wenner 四极法测量电阻率,考虑到修正电导池常数取决于试样的几何形状,将电阻率的结果转换为材料特性。通过阿基米德法测量混凝土试样的孔隙率,使用压汞法测量小试样的孔隙率。 模拟 将 CT 扫描获得的图像数据导入 Simpleware 软件,基于阈值分割为硬化水泥浆体和孔隙两个区域,分界线根据压汞法和阿基米德法测得的孔隙率确定。 图1:灰度像素值直方图和 3D 孔隙结构模型 在 Simpleware FE 模块生成高质量的四面体网格模型,导出至 COMSOL 软件进行数值模拟。在 X 方向施加电位差,计算 X 方向每个平面上观察到电流密度的面积分,从而得到流过多孔介质的平均电流 I。绝缘平面的边界条件设定为诺伊曼条件,根据稳态模拟结果可根据电位 V 和电流 I 得到电阻率 R。 为确认结果各向同性的一致性,在 X、Y、Z 方向上均进行计算。值得注意的是,液相中的电导率很大程度上取决于孔隙溶液中的离子浓度和迁移率,满足电中性的要求。电流通过离子传导在硬化混凝土中流动,在模拟中假设其是完全饱和的。 […]

概述 众多可持续发展目标需要探索和生产新的可持续材料。作为最有前途的材料之一,多孔金属结构由于其较高的表面积、刚度和孔隙体积及固定的孔隙网络成为改善流体和传热的理想候选材料。固体含量较高的泡沫为闭孔金属泡沫,而固体含量较低的泡沫为多孔金属泡沫。材料的拓扑和孔隙结构特征往往受到生产工艺(发泡、铸造、烧结等)路线和操作条件的影响,可由几乎所有液态金属或金属粉末制成,包括铝、铜、镍、钢、铁和合金。多孔金属可广泛应用于航空航天、热力水力输送、燃料电池、吸声板、空气净化技术和环境减排等。 为设计可应用于高效传热传质的金属泡沫,了解其流体结构特性、流体流动状态和边界是非常必要的。本项目采用多学科方法,利用实验、计算流体动力学(CFD)建模和仿真以及人工神经网络(ANN)机器学习反向传播研究液态熔体渗透技术制备铝泡沫的流体动力学。 图像处理 将含 99% 铝的液态熔体加热至 800 ℃ 后分别倒入由近球形盐、软水盐和粒状盐的空心填充床组成的模具中,凝固后压实。 图1:由近球形盐(1.4-2.0 mm,a2)和粒状盐(3.0-4.0 mm,b2)制成多孔铝结构的摄影图像(左)和扫描电子显微镜(SEM)图像 使用 Zeiss Xradia Versa XRM-500 X 射线计算机断层扫描(CT)系统获得图像数据集(3000 张 TIFF 格式),导入 Simpleware 软件中进行图像处理,应用各种工具(阈值、滤波器、腐蚀和膨胀等)生成 3D 体积结构。在Simpleware ScanIP 模块中,从一个大的 3D 模型中裁剪出合适的 3D RVE(代表性体积单元)结构,使其测量的孔隙率与实验得到的名义孔隙率仅相差 ±3 %,尺寸为材料平均开孔直径的 3-5 倍。在 ScanIP 中可以直接测量铝泡沫的孔隙率、体积和表面积,平均孔径和平均开孔通过平均分水岭分割孔隙和开孔流体域 3D RVE 的中心线得到。 在 Simpleware FE 模块生成 3D RVE 流体域的四面体网格模型,最小和最大边长分别为 3 倍和 7 倍体素尺寸,获得 5 种铝泡沫试样的最佳网格密度(2.6-3.1 Mcells)。设定边界条件,求解 […]
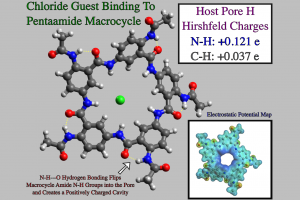
如果主体对携带特定电荷的客体物种具有亲和力,则可能用于对于对抗各种疾病。合成的超分子结构被设计为与阳离子或阴离子结合,利用官能团的影响得到大环的孔,以获得负电荷与阳离子相互作用,或正电荷以结合、运输阴离子。通过迫使孔隙带正电,让生物膜主体具有阴离子结合特异性,这对于对抗氯离子输送故障和治疗囊性纤维化非常重要。在最近的一项研究中,主体五酰胺大环被特异官能化,通过N-H–O氢键迫使酰胺基团被定向到孔中,如此将相当大的正电荷放入五酰胺的空腔中,并将羰基翻转到大环之外。由于孔中 NH 和 CH 的正电荷,这些五酰胺大环对阴离子具有特异亲和力。 五酰胺大环上的 Hirschfeld 电荷和静电势,以及单体构建块的偶极矩,使用 ADF 计算得到,证明了腔的强正电性。几何优化表明,过量阴离子客体被吸引到五酰胺大环的正电空腔中。计算的缔合常数和结合能表明,主体空腔偏向去溶剂化,以允许孔内容纳阴离子客体。本文特别关注了主体结构内氯化物、溴化物和碘化物客体的结构优化,计算表明碘化物与五酰胺腔的结合最强,其次是氯化物,溴化物的结合最弱。这与 N-H–Cl– 相互作用强于 N-H–Br– ,N-H–I– 相互作用最弱的一般趋势相反。结构优化表明,碘化物与五酰胺大环的结合最强,因为它的尺寸更大,能够与孔中的所有N-H基团相互作用。由于氯化物和溴化物的尺寸较小,这两种阴离子只能聚集两个 N-H 基团配位。考虑到氯化物比溴化物的结合力强,又不像碘化物那样过度填充孔隙,因此相对于其他卤化物,氯化物的传输能力最好。 因此,正如ADF计算结果所显示的那样,五酰胺大环是对抗囊性纤维化和补充氯化物正常运输的理想选择。 参考文献 Ruikai Cao, Robert B. Rossdeutcher, Yulong Zhong, Yi Shen, Daniel P. Miller, Thomas A. Sobiech, Xiangxiang Wu, Laura Sánchez Buitrago, Karishma Ramcharam, Mark I. Gutay, Miriam Frankenthal Figueira, Pia Luthra, Eva Zurek, Thomas Szyperski, Brian Button, […]
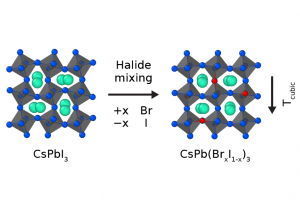
摘要 金属卤化物钙钛矿已成为光电应用中极具前景的材料,其卓越的性能使其成为太阳能电池和LED的理想候选材料。现有的钙钛矿组合物通常涉及不同离子的混合,目的是针对特定应用,对材料的光电性质和稳定性进行微调。为了理解离子混合的原子效应,埃因霍温理工大学和宾夕法尼亚州立大学的研究人员开发了一种用于大规模分子动力学模拟的无机金属卤化物钙钛矿(CsPbX3,X=Br 或 I)的 ReaxFF 力场[1]。 研究人员利用 DFT 计算生成的参考数据集,使用 AMS 软件中的 ParAMS 参数化功能,基于先前开发的 CsPbI3 的力场(该力场有助于研究体材料降解反应[2]以及表面和晶界的影响[3]),对该力场进行了扩展, 得到了新的无机卤化物钙钛矿的 ReaxFF 力场[4] CsPb(BrxI1-x)3 ,新开发的 I/Br/Pb/Cs 参数集首次对 Br 进行描述。在包括状态方程、混合焓、降解反应和缺陷迁移能垒在内的各种基准测试中,该力场表现良好。在分子动力学模拟中,该力场可以准确地再现材料的有限温度效应,例如无机钙钛矿的各种体相之间的相变。 使用新的力场参数,研究人员确定,由于 I 和 Br 离子的尺寸失配,卤化物混合对材料中八面体的相变温度和倾斜动力学有着深远的影响。利用卤化物混合的稀释极限(即取代钙钛矿晶格中的单个卤化物),确保这种效应是非局部的,距离混合位点高达两纳米。混合效应的非局部性,解释了为什么少量卤化物混合会对材料性质(如相变温度)产生很大影响。新的 ReaxFF 力场参数,为大型现实体系通过原子模拟,进一步探索无机混合卤化物钙钛矿的复杂动力学铺平了道路。 参考文献 Pols, M.; van Duin, A. C. T.; Calero, S.; Tao, S. Mixing I and Br in Inorganic Perovskites: Atomistic Insights from Reactive Molecular […]
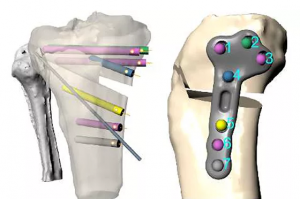
概述 治疗早期膝骨关节炎的其中一种方法是胫骨高位截骨术(HTO)。然而,HTO 具有挑战性且可能引起并发症。在本研究中,通过使用 Simpleware 软件创建 28 名患者的虚拟模型开发了一种新的个性化 HTO 器械,采用计算机模拟试验与标准治疗比较,验证的骨科器械的安全性。 结果表明,该个性化器械与标准器械具有一致的安全性,已申请在英国获得 MHRA 批准,应用于正在进行的临床试验,增加 HTO 作为治疗选择的使用。 亮点 为比较个性化 3D 打印胫骨高位截骨(HTO)器械与现有通用器械的机械安全性进行计算机模拟试验。 使用 Simpleware 软件处理患者 CT 数据和通用器械的扫描图像,生成 28 个可用于模拟的准备模型。 结果表明,新型器械的安全性并没有增加风险,可为个性化 HTO 提供支持。 介绍 胫骨高位截骨术(HTO)是一种简单有效治疗早期膝关节骨性关节炎的方法。相较于全膝关节置换,年龄较小的患者可能更倾向于 HTO。而全膝关节置换在老年人中更常见,因为他们对长期性能的需求较小。然而,由于难以实现精准的计划矫正,很少有外科医生考虑这种手术。在本案例研究中,通过计算机模拟临床试验比较新型个性化 3D 打印 HTO 板和现有通用模型,更好地评估安全性和有效性。 胫骨高位截骨术是在胫骨近端创建一个开口或闭合的楔形截骨,改变内翻对线,从而改变小腿的机械轴线,减少疼痛间室的负荷。截骨术通常使用接骨板稳固,常见的并发症包括疼痛和软组织刺激引起的不适。因此,实现足够的钢板柔韧性对于长期骨愈合和积极的患者预后至关重要。将针对于特定患者的接骨板优化为独特的骨骼几何形状,可能会提高其适配性和减少并发症。此外,具有个性化胫骨几何形状的数字化三维规划可以在术前确定螺钉的长度和方向,有助于减少手术时间。 在计算机模拟临床试验中,可以在同一个体的虚拟模型上预演多个手术,配对比较新型和已建立干预措施之间的力学结果和失败风险。这些试验遵循与物理临床试验相同的惯例和对照方案。本研究特别关注通过有限元分析(FEA)得到的生理负荷期间植入板上机械应力峰值。 图像处理 对 30名膝骨关节炎患者进行 CT 扫描,其中 2 名患者因 CT 扫描结果不佳而不纳入考虑。使用实验测得的刚度和强度变化对标准尺寸 TomoFix HTO 板(DePuy Synthes)进行功率分析,有助于确定患者队列规模的适宜性。Tomo Fix HTO 板是一种广泛植入的 HTO器械,在本研究中作为“通用器械”。 将 CT 数据导入 Simpleware 软件进行分割,确定 5 个感兴趣的关键标志点。利用 MATLAB 计算出所需的截骨矫正角度,使改变的机械轴穿过从内侧到外侧胫骨平台距离 62.5 % 的点。对每位患者进行虚拟 HTO 手术,通过创建开口楔形截骨改变膝关节的机械轴,手术模拟在 Ansys SpaceClaim 中进行,由专门从事膝关节手术的骨科医生指导。 图2:采用由 Simpleware 软件创建的 3D 几何结构在 Ansys […]

传统的气体压缩式制冷技术由于能耗大、效率低、环境不友好等问题急需进行变革,而磁制冷技术作为一种基于磁热效应的新型固态制冷技术,有望解决传统制冷技术面临的一系列问题。近年来,La(Fe,Si)13金属间化合物(结构如图1)由于其在一阶相变过程中的巨磁热效应,成为最有前途的室温磁制冷材料之一。然而,一阶相变伴随的热滞和磁滞会导致制冷效率的降低。因此,科研人员致力于通过掺杂来解决这一问题。
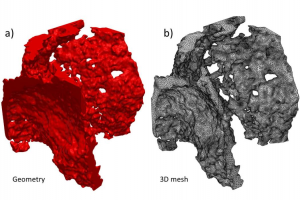
概述 钢铁产量中近 70% 来自采用高炉(BF)工艺生产的生铁,因此钢的成本很大程度上取决于 BF 的寿命。由于耐火材料的劣化,BF 的寿命有限,特别是聚集着液态金属的高炉炉膛中使用的耐火材料。设计更具有适应性的新型耐火材料需要全面了解工作过程中发生的劣化机制,而各种机制都取决于耐火材料的孔隙结构,并受到多孔介质中传输的限制。 本项目基于 X 射线计算机断层扫描(XCT)获得 3D 孔隙结构,开发并测试了一种耐火材料中熔融金属渗透的演化模型。假设等温渗透,材料中存在的相具有不同的润湿性且碳相选择性溶解,分析不同初始熔融金属成分和各种润湿条件对渗透过程演变的影响。 试样准备 微孔碳材料由 Tokai COBEX 公司生产,主要原料是人造石墨粒、人造半石墨粉、硅粉和氧化铝粉,煤焦油沥青为粘结剂。固体颗粒与粘结剂的比例为3.64:1。将混合原料成型为 2500 × 700 × 500 mm3 的块体,在标准环形炉内还原气氛下烘烤,从制成样品中切割直径和高均为 10 mm的圆柱体作为试样,采用 XCT 进行分析。 准备三种类型的基材用于润湿性测量:石墨(G)、氧化铝(A)和碳化粘结剂(B),全部由用于生产微孔碳材料的相同原材料制备。为获得样品的各种化学成分,准备生铁废料、化学纯铁、工业纯铁、化学纯锰和两个对照熔体,分别表示为:M2C、M3C 和 M4C。为验证化学成分,在每次熔炼后使用 Foundry-Master 光谱仪分析铸铁样品的化学成分。 表1:用于润湿性测量准备的粗铁成分(wt%) 数据处理 使用 Nanotom 180S 设备(GE)进行 XCT 扫描,将原始图像数据裁剪为 1.25 × 1.25 × 1.25 mm3 的立方体,导入 Simpleware ScanIP 软件进行图像处理。基于灰度值分割为 5 个不同的相:碳化粘结剂(棕色)、石墨(蓝色)、氧化铝(绿色)、开孔(红色)和闭孔(黄色),其中使用 Flood Fill 3D 算法工具识别开孔。 图1:XCT 数据处理过程:(a)实测数据(b)裁剪数据(c)分割数据 在 Simpleware FE 模块采用 […]
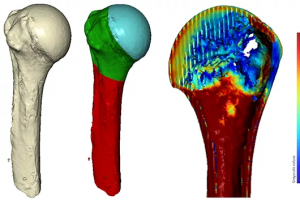
概述 随着人口老龄化,骨质疏松越来越受到关注,导致骨折和手术并发症的风险增加。为做出明智的治疗决策并识别容易出现骨密度相关问题的患者,外科医生需要在手术前了解患者的骨质量。然而,目前的骨密度分析方法并不十分可靠和准确。 本研究旨在开发一种术前根据 CT 数据测量肱骨近端局部松质骨矿物质密度(BMD)的方法。研究人员通过 Simpleware 软件使用感兴趣三维区域的方式,比现有方法更精确和一致。 亮点 术前利用 CT 数据测量肱骨近端局部松质骨矿物质密度的方法。 通过使用 μCT 数据对标准 CT 扫描进行准确性分析验证测量结果。 Simpleware 软件可以确认骨密度的空间位置(可靠性),并从临床 CT 扫描中准确提取骨密度特征。 图像获取 通过临床 CT 扫描设备(Siemens SOMATOM Definition AS+)扫描 30 具尸体肱骨,使用制造商的密度模型将灰度值转换为 BMD [mgHA/cm3] 值。额外的 microCT(μCT,Phoenix v/tome/x s)扫描,体素分辨率为 50μm,BMD [mgHA/cm3]中已校准体模 82 灰度值作为评估密度分析准确性的参考。 分析方法 图2:验证3D CT 骨密度测量的分析方法 使用 μCT 数据在体模校准队列(n = 30)的标准 CT 扫描中进行准确性分析。通过自动图像处理脚本为密度分析进行可靠性分析。 使用临床 CT 数据的组内相关系数(ICC)和图像处理脚本与 μCT 数据和 Simpleware 工具性分析评估准确性和可靠性。对单个测量值进行双向随机效应分析,并将可靠性应用于单个评分者/方法的单个测量值一致性的背景下。ICC 大于 0.75 为优秀,0.40 […]

概述 固体氧化物燃料电池(SOFC)是一种极具吸引力的电化学装置,可以直接有效地利用燃料的化学能发电。SOFC 在相对高温(600–1000 ℃)下工作,可实现较高的燃料灵活性,并通过热电联产系统中的废热回收提高效率。为充分发挥潜力,SOFC 需要进一步改进,在经济上与传统能源转换技术竞争,包括提高其耐用性和可靠性。 渗透是在预制电极骨架内生成具有特定性质(如特定电导率或电催化作用)纳米级固体的过程。渗透改变了电极的局部形态,对电化学反应位点的数量和/或质量和电荷传输具有积极的影响。在实验上,很难直接确定局部电化学如何受到影响和在相关长度尺度(几十微米量级)上以适当的分辨率(纳米量级)量化电化学活性位点的 3D 分布。 为了解纳米级渗透如何影响性能,需要能够模拟许多相对较大体积 3D 微观结构的计算工具。本项目通过研发的高通量开源仿真代码 ERMINE 对 55 种不同的阴极微观结构进行有限元模拟,探索 SOFC 中离子导体和电子导体作为渗透物的差异。 图1:从分割图像数据到模拟的计算工作流程 亮点 对渗透 SOFC 阴极进行电化学 HPC 模拟。 离子导体的渗透比电子导体更能提升性能。 离子导体将在 TPB 处产生的电流重新分布到整个电极上。 再分布降低了 TPB 处的局部欧姆过电势,增加了局部活性。 原始微观结构更能显著影响整体性能。 图像处理 采用商业阴极结构的图像数据进行 3D 重建,原始数据尺寸为 126 × 73 × 12.5 μm3,体素为 55 × 55 × 50 nm3,基于灰度值将其分割为氧化钇稳定的氧化锆(YSZ)、镧锶锰氧化物(LSM)和孔隙。从完整模型中随机提取五个尺寸为 10 × 10 × 7 μm3 的较小微观结构子体积,记为 BB_i (i = 1-5)。为更精准地分割出三相界面(TPB),通过重采样调整分辨率,将原有的 1 个体素由 8 个相同的更小体素组成。 对五个微观结构进行人为地渗透,随机选择与 LSM 和 YSZ 表面接触的一定比例孔隙种子。将渗透相(LSM […]