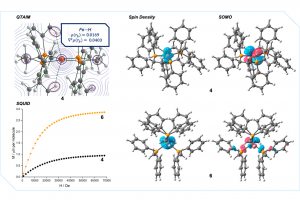
研究背景 寻找对现有试剂的低成本、低毒的替代品,一直是均相催化领域的核心诉求。近年来,利用地壳富含的铁元素构建催化剂来取代昂贵且不可持续的贵金属元素,已成为该领域的研究热点。其中,一价铁(Fe(I))分子配合物更是在诸多工业催化反应中展现出优异的催化性能。然而,性能优势的背后暗藏不可忽视的瓶颈。与第9、10族贵金属催化剂不同,低价铁面临着一个致命缺陷——缺乏化学性质稳定的起始物料。传统合成方法只能高度依赖钾石墨(KC8)或格氏试剂(RMgX)等强还原剂,对二价或三价铁前驱体进行原位还原。这不仅导致催化体系结构不明确,还极大地限制了反应的官能团兼容性。尽管此前学界曾尝试通过氧化铁羰基化合物或高价铁化合物还原法制备双夹心铁芳烃配合物,但往往因发生歧化反应难以分离得到纯净产物,最终功亏一篑。针对这一痛点,Oliver P. E. Townrow 等人在国际顶尖化学期刊 JACS 上给出了破局之法,课题组成功开发出一种在固态下对空气稳定的单核一价铁夹心配合物 [Fe(durene)2]+(durene = 1,2,4,5-四甲苯)。这是首个稳定的一价铁合成单离子“源头”!作为一种前所未有的稳定前驱体,只需在外加配体的存在下,它就能通过温和、氧化还原中性的配体交换反应,模块化地构建出一系列已知及全新的一价铁化合物,彻底摈弃了对强还原剂的依赖。作为概念验证,团队进一步证实该单离子源可在原位转化为高活性的 Kumada 交叉偶联催化剂。这一突破不仅为低价铁的高通量筛选铺平了道路,更为实现丰产元素催化的工业化应用搭建了一个全新的通用型平台。 图1 工作背景与灵感来源 研究内容 一、合成与表征 针对传统制备方法易过度还原且产物极不稳定的问题,本研究开发了一条选择性合成一价铁单离子源的新路径(图 2)。该路径以二价铁配合物为起始原料,创新性地引入有机还原剂 TMS2Pz* 进行还原,实现了高度专一的单电子还原,即使使用过量还原剂也不会生成零价铁副产物。随后,采用 NaBArF24 的 DCM 饱和溶液引入弱配位阴离子 [BArF24]–,有效稳定了溶液中的低价铁离子中心,并最终以高达 75% 的结晶产率成功制备出可进行数克级规模合成的目标产物 [Fe(durene)2][BArF24] (3Dur)。该合成路线不仅彻底摆脱了对强还原剂的依赖,还赋予了产物前所未有的稳定性:3Dur 可在空气中轻松处理与称量,室温下24小时内无明显氧化迹象,真正实现了实用型一价铁单离子源的高效制备。此外通过多种谱学与磁学表征明确了3的结构(图2):即 3Dur 呈 S=1/2 低自旋基态;化合物3的芳环间呈交错构象;3Mes 结晶于 P21/c 空间群,其不对称单元中包含一个离子对;3Dur 结晶于 P21/n 空间群,不对称单元中包含两个结晶学不等效但结构相似的离子对以及一当量的氟苯;3Bz* 结晶于P21/c 空间群,含有一当量的二氯甲烷。以上三者之间的铁-芳环质心距离保持一致,且该距离随取代基的增加而变长。电化学分析表明,与强还原性、易活化催化剂配体的传统Fe(I)夹心化合物不同,结构 3 的 Fe+/2+ 氧化电位显著更正,使其显著适合作为稳定的一价铁单离子源。 图 2 [Fe(durene)2][BArF24] (3Dur) 的合成方法,180 K 下通过单晶 X 射线衍射测定的 3Mes、3Dur […]