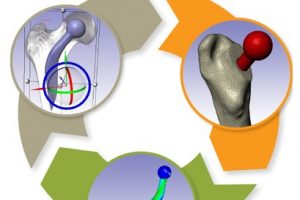
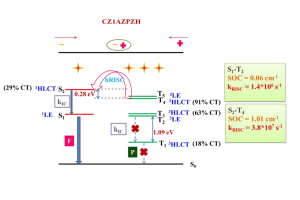
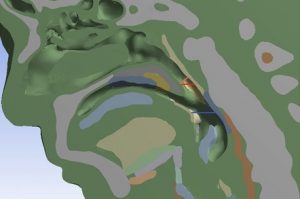
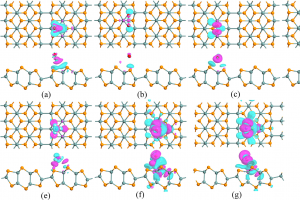
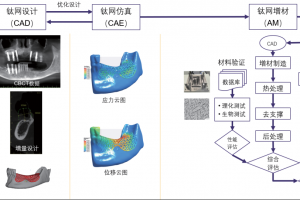
概述 植入物定位是全髋关节置换术的一个重大挑战。植入物必须要很好的适合并放置在髓管内,尽量使股骨—植入物的接触面积最大化。然而植入物位置的实验测试会超出成本的限制。另一种选择是使用计算模型在产品研发初期就综合分析植入物的位置。尽管这种方法并不是为了取代实验,但它可以帮助外科医生更好地理解植入位置对原发或继发稳定性的影响。结合 Simpleware 软件与 ANSYS 创建自动化的工作流程,整合 CAD 设计的植入物和股骨 CT 扫描,生成用于微动分析的有限元模型。模拟结果生成的响应面证实了位置变化对微动的影响。 亮点 在Simpleware软件中整合CAD植入物和股骨CT扫描在Simpleware软件中生成FE网格通过脚本自动生成多组植入物位置/方向在ANSYS Workbench中进行植入物微动的模拟对结果进行后处理并生成响应面预测植入物位置的最佳和最差情况 图像处理和CAD整合 使用Simpleware ScanIP和Simpleware CAD将由CT扫描进一步处理获得的分割后的股骨模型与CAD设计的植入物结合。然后利用Simpleware FE生成有限元网格,导出至ANSYS Workbench中进行微动模拟。通过Simpleware API运用Python脚本自动生成多个植入位置,而无需耗费大量时间手动调整。 图:使用Simpleware CAD将分割后的股骨与CAD设计的植入物结合 FE网格生成 在本例中,为每个植入位置生成有的限元网格包含股骨的约10000个节点和38000个单元,钛金属植入物模型约2000个节点和6000个单元。采用Simpleware软件的自动转换算法基于原始扫描的CT值(Hounsfield单位,HU)为股骨分配标准材料属性。为模拟约束和加载条件给植入物和股骨添加节点集,在植入物—骨界面处的网格细化也增加了模拟的真实性。 图:使用Simpleware FE对股骨和植入物进行网格划分 应力分析&响应面模型 将初始的有限元模型导出至ANSYS Workbench,在上千个可能的候选的基础上产生成功的微动模拟。利用Kriging回归法对425个成功模拟点进行插值,生成响应面模型(RSM)。 ANSYS Workbench模拟可以利用RSM确定导致微动最高和最低可能值对应的植入位置。外科医生可以根据这些结果解释和预测植入物的最佳和最差位置。 图:在ANSYS Workbench中获得的最佳位置(左)和最差位置(右) 结果:最佳/最差的植入位置 利用Simpleware软件和ANSYS结合图像数据与有限元分析,成功地开发了一种用于分析植入物与股骨间相互作用的自动化工具。采用的响应面方法使人们深入了解微动对定位的敏感性。 随着该工具可行性的建立,进一步的工作可以集中在研究多个植入物设计在患者群体的分析。因此除了植入物研发之外,该工具在手术规划方面也有非常重要的应用。 参考 致谢和更多信息请参考英文原文:https://www.synopsys.com/simpleware/resources/case-studies/total-hip-replacement.html。