
研究背景
在纳米材料科学的前沿领域,原子级精确的贵金属分子团簇因其卓越的稳定性和明确的结构,被视为构建新型功能材料的理想基石。通过引入阴离子作为模板剂,引导金属纳米团簇形成独特的笼状空间构型,并以此为平台深入探究“结构-性能关系”。然而,相比于研究较深入的金纳米团簇,铜与银团簇,尤其在阴离子封装特性上的对比研究仍显不足。近日,知名学者 Alvaro Muñoz-Castro 在无机化学领域权威期刊 Inorganic Chemistry 上发表了题为 “Copper and Silver Cuboctahedron Cavities in Halide Encapsulation: Evaluation of Interaction Energy and Optical Properties Given by the Inclusion of Halides as Non-Innocent Templates” 的研究论文。该工作以具有立方八面体空腔的同构团簇 [M12(μ12-X){S2P(OnPr)2}6{C ≡ CPh}4]+(M = Cu, Ag; X = Cl–, Br–, I–) 为研究模型,系统评估了不同卤素阴离子与金属笼之间的相互作用本质。研究发现,银笼相比铜笼具有更大的空腔可调性与更小的空间位阻;通过能量分解分析(EDA)同步揭示了静电作用在稳定封装中的主导地位,并证实了阴离子在调控团簇前沿轨道与光学性质中的“非惰性”角色。这一成果不仅深化了对金属宿主与内嵌阴离子间非共价相互作用的理论认知,也为未来理性设计面向催化、传感及分子识别领域的定制化纳米材料提供了重要指引。
研究内容
1,结构特征
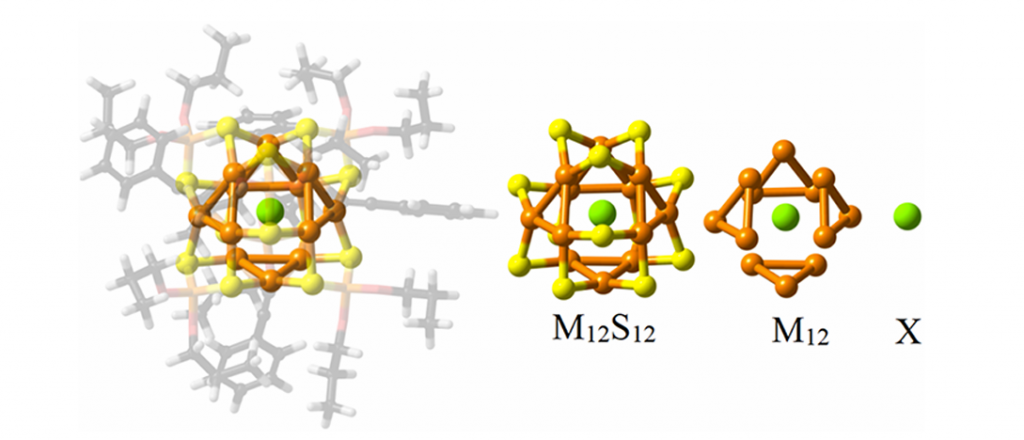
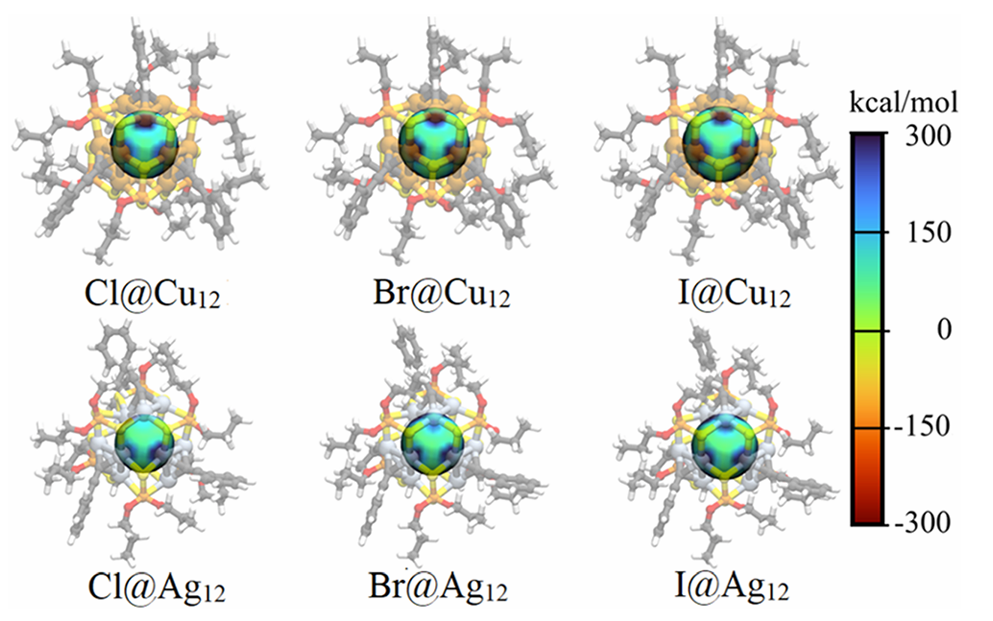
通过对 [M12(μ12-X){S2P(OnPr)2}6{C ≡ CPh}4]+(M = Cu, Ag; X = Cl–, Br–, I–)系列团簇(图1)的几何结构与拓扑对称性分析,揭示了铜、银金属笼在封装不同尺寸卤素阴离子时的自适应结构重排规律。理论计算表明(表1),空心母体笼的初始半径分别为 Cu12(2.860 Å)和 Ag12(3.327 Å);在封装卤素时,较小的 Cu12 空腔被迫显著扩张以容纳内嵌原子(其半径计算值从 Cl– 的 2.980 Å 增大至 I– 的 3.120 Å,与实验趋势一致),而 Ag12 空腔半径变化相对较小,表明其内部空间充裕。同时在 Cu12 空腔中,引入卤素后其半径呈现为逐渐增加的趋势;而在 Ag12 空腔引入半径较小的 Cl– 和 Br– 时反常发生内向收缩,但引入 I– 时则会增加,展现出比铜笼更优异的空间宽裕度与尺寸调节能力。此外,利用连续形状测量(CShM)方法对金属笼多面体构型进行评估,结果表明无论是空心笼还是含卤素离子团簇,M12 空腔结构均更接近于立方八面体而非二十面体,且随着包裹的卤素由硬路易斯碱(Cl–)向软路易斯碱(I–)演变,立方八面体的畸变程度逐渐增大。
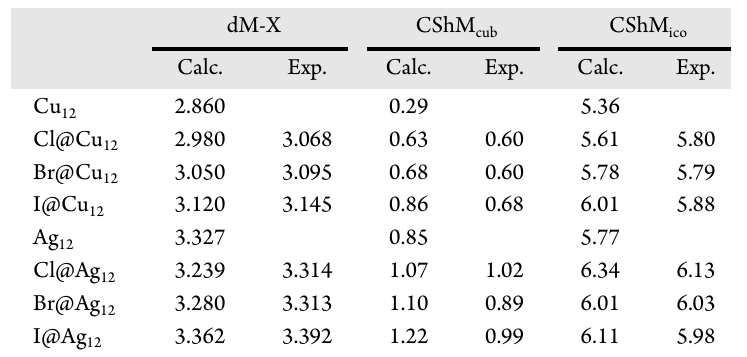
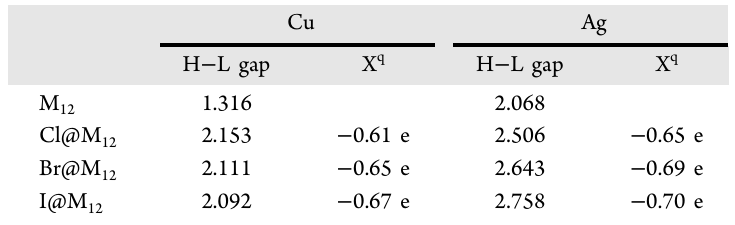
由于内嵌卤素的 npX 轨道与金属笼之间存在反键相互作用,团簇的 LUMO 能级去稳定化,进而使得 Cu12 系列和 Ag12 系列的 HOMO-LUMO 能隙分别增大至 1.316–2.153 eV 和 2.068–2.758 eV,其中 Cl@Cu12 和 I@Ag12 表现出较大的前线轨道能隙,这揭示了卤素在两种金属笼中对最终能隙的影响存在显著差异。自然布居分析(natural population analysis,NPA)显示,封装卤素上的电荷在 Cu12 团簇中为 -0.61 至 -0.67 e,这对应 0.39 至 0.33 e 的电荷转移发生于 X→M12间;而在 Ag12 物种中,卤素电荷为 -0.65 至 -0.70 e,净电荷转移量减少为 0.35 至 0.30 e,这表明相比于铜团簇外壳,银团簇外壳表现出较弱的电荷接受能力。
通过计算预备能(ΔEprep)以量化 Cu12 和 Ag12 团簇容纳卤素所需的结构畸变的能量成本。结果表明,Cu12 团簇的 ΔEprep 随卤素体积增大而增加(Cl@Cu12 为 2.3 kcal/mol,I@Cu12 增至 5.8 kcal/mol),这体现了铜团簇的整体柔性,而 Ag12 团簇的 ΔEprep 变化较小且在 I@Ag12 结构时最低(0.8 kcal/mol),结果反映出不同金属笼对卤素尺寸的偏好差异,且这种变化主要源于 M12 核心而非外部配体。相互作用能(ΔEint)的分析表明 Ag12 笼的卤素离子封装比 Cu12 更有利,基于 M122+-X– 片段划分模型的 Ziegler-Rauk 能量分解分析,银笼的卤素封装更为有利(-154.9 (Cl@Cu12) > -151.5 (Br@Cu12) > -148.2 (I@Cu12) kcal/mol,-167.8 (Cl@Ag12) > -160.5 (Br@Ag12) > -158.1 (I@Ag12) kcal/mol),其中静电相互作用(ΔEelstat,平均占比75.6%)是绝对的主导稳定化因素,其次是轨道相互作用(ΔEorb,平均占比 20.2%)与色散作用(ΔEdisp,平均占比 4.2%)。
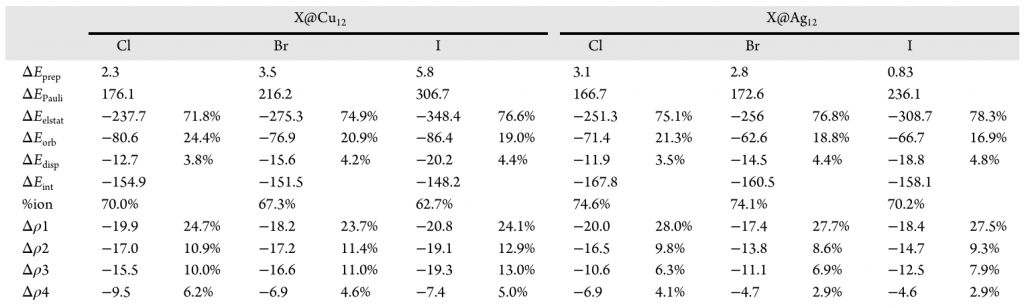
通过分子静电势分析及与稀有气体模型对比,阐明了团簇的静电作用的位点及其离子性特征。随着卤素离子从 Cl– 到 I– 范德华半径的增大,静电贡献显著增强。利用稀有气体(Ng)等电子体替代模型量化离子贡献(ion%)的结果显示,静电项中的离子特征占比随卤素体积增大而降低(Cu12体系中由 70.0% 降至 62.7%),表明偶极-偶极等极化作用的贡献逐渐增加。这意味着 Cu12 和 Ag12 笼相比有机主体提供了更具极化性的封装环境,根据 HSAB 理论,这种环境赋予了相互作用更软的特征,从而揭示了空腔特性对静电相互作用有效的调节作用。此外,随卤素半径增大急剧上升的泡利排斥能(ΔE Pauli)的数值在 Ag12 体系中显著低于 Cu12 体系,再次印证了银笼内部空间位阻更小的特征。
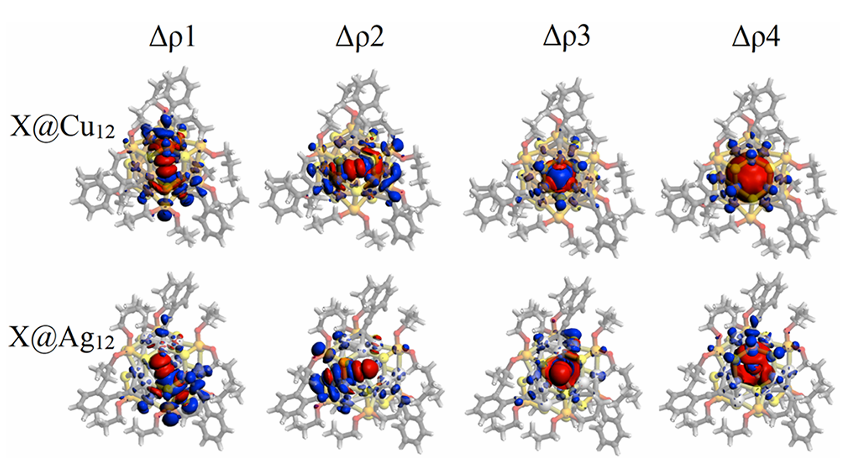
通过 EDA-NOCV 分析进一步将轨道相互作用细化为四条电荷转移通道(三个 npX→M12 和一个 nsX→M12),从而在空间和能量尺度上完整呈现了阴离子@金属笼复合物的精细配位图景。
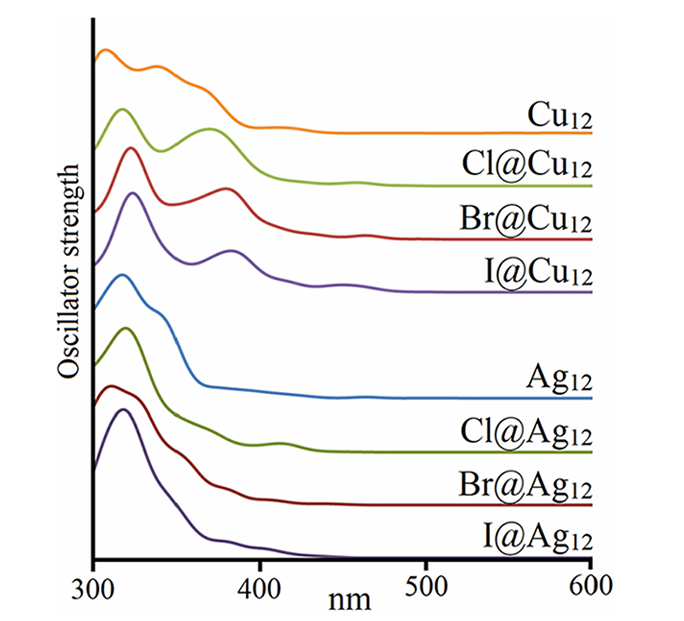
最后,进一步评估了卤素离子嵌入对金属笼光学性质的影响。结果表明,X@Cu12 和 X@Ag12 系列的 UV–vis 光谱仅显示出微小差异,表明两者具有相似的前线轨道特征。计算结果显示(考虑 TD–DFT 约 0.2–0.3 eV 的典型误差),X@Cu12 物种的第一个主峰位于 378–381 nm(Cl、Br、I依次变化),与实验测得的 332、332 和 331 nm 信号吻合良好,同时计算出的次要主峰位于 317–318 nm,均与实验谱图相符。相比之下,银基物种的第一个主峰出现在 318–320 nm 处,对应于实验观测到的 290–292 nm 信号,呈现出与
X@Cu12物种双主峰模式不同的光谱特征。
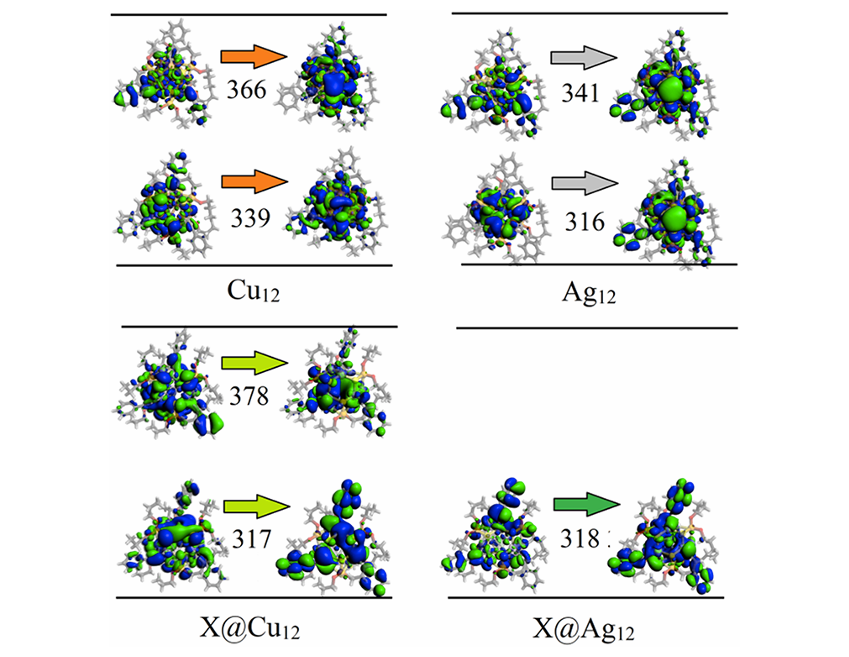
自然跃迁轨道(NTO)分析表明,铜和银系列中的相关跃迁主要涉及配体和 M12 簇核心的电荷转移跃迁,其中高能信号表现出更强的配体特征。这一结果与 Ag12(μ12-X)(μ3-I)4(S2PH2)6+ (X = Cl, Br, I) 簇中观察到的 LMCT 和 XLCT 跃迁相似,但相较于 X@Cu12 和 X@Ag12 系列,这些跃迁发生了蓝移,从而凸显了外部配体在其中的重要作用。
总结与展望
综上所述,本研究以高度对称的同构纳米团簇 [M12(μ12-X){S2P(OnPr)2}6{C≡CPh}4]+(M = Cu, Ag; X = Cl–, Br–, I–)为原型,系统地阐明了立方八面体贵金属笼在封装不同卤素阴离子时几何柔性、电子结构及非共价相互作用之间的协同效应。结果表明,相比于小尺寸且空间位阻较大的铜笼,银笼展现出更宽敞且更具柔性的自适应空腔,能更高效地容纳大尺寸卤素;而能量分解分析(EDA-NOCV)则进一步证实,这种强烈的封装相互作用主要由静电作用(约 75.6%)驱动,并辅以轨道相互作用(约 20.2%)与色散力(约 4.2%)的稳定化贡献。不仅如此,内嵌卤素作为“非惰性”模板剂,其离子特征不仅遵循软硬酸碱(HSAB)理论随原子序数增加而减弱,更通过特定的供体-受体通道引起了团簇前线轨道和紫外-可见吸收光谱的系统性调控。这一成果不仅深化了研究者们对内嵌阴离子金属宿主结构基本物理化学性质的理论认知,也为未来宏观上理性设计并构筑具有特定光电性能、高稳定性的可调控纳米宿主材料提供了清晰的理论边界,对推动其在催化、高灵敏传感以及精准分子识别等前沿材料科学领域的创新应用具有重要的指导意义。
参考文献
- Paco-Chipana M, Muñoz-Castro A. Copper and Silver Cuboctahedron Cavities in Halide Encapsulation: Evaluation of Interaction Energy and Optical Properties Given by the Inclusion of Halides as Non-Innocent Templates. Inorganic Chemistry, 2026. DOI: 10.1021/acs.inorgchem.5c05316