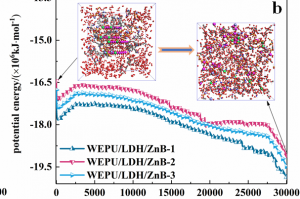
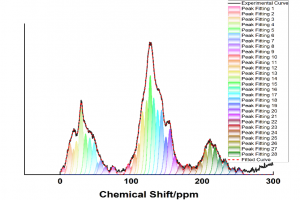
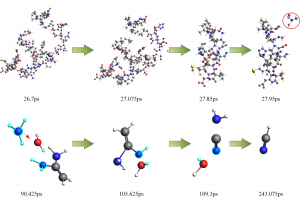
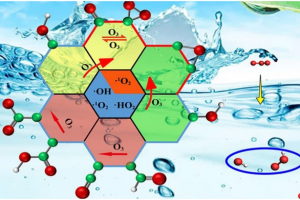
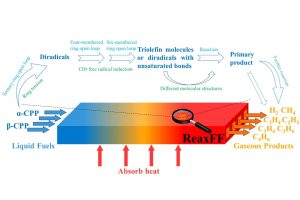
研究背景 发泡聚苯乙烯泡沫具有保温、吸水、抗压减震、耐候性好等优点,被广泛应用于产品包装、建筑消防、化工生产、汽车工业和航空航天等众多领域。根据相关统计,普通 EPS 阻燃性能较差,暴露在明火中容易发生分解燃烧,且燃烧时伴随着大量的浓烟和刺鼻气体,对人体和环境构成巨大威胁。因此,提高 EPS 的阻燃和抑烟性能至关重要。 研究内容 本研究由沈阳理工大学和辽宁工程技术大学等单位合作,基于机器学习势方法,使用 AMS 软件中的 ML Potential 模块完成燃烧模拟工作。构建铝镁水滑石、硼酸锌、聚氨酯和环氧树脂阻燃涂层晶胞模型。通过模拟手段从微观层面对涂层体系进行燃烧反应研究,该方法不仅能够直接获取涂层结构体系的燃烧演化过程,还能够利用微观阻燃机理揭示宏观阻燃现象。 图1 涂层分子模型 在 15000 fs 内,三种涂层体系内的总分子数量随温度升高而快速增加;15000-27500 fs 内,分子数量发生小幅度上下波动,说明燃烧逐渐减弱,分子的消耗和生成逐渐趋于平衡;25750-30000 fs 内,分子数量随温度降低而逐渐减少。0-2500 fs 阶段,涂层体系内势能的增加表明发生吸热反应;2500-27500 fs 阶段,体系内势能缓慢降低;27500-3000 fs 阶段,体系势能随温度降低而快速减小。 图2 燃烧过程中体系分子数量和能量的变化规律 图 3 列举了涂层体系燃烧后留下的主要金属化合物和硼氧化物。这些金属产物所含原子数量不同,小的不足 10 个,大的在 35 个以上,分子结构中包含 Zn、B、Mg 和 Al 金属原子。WEPU、LDH 和 ZnB 分子结构在燃烧反应过程中主要通过 O 原子促使分子链之间相互连接,形成新的金属化合物片段。这些金属化合物会形成一层覆盖在基材表面的固态物质,能够有效阻隔热量向基材内部传递,减缓基材的升温速度,阻止氧气与基材直接接触,从而抑制燃烧反应的持续进行,保护 EPS 基材不被进一步燃烧破坏。 图3 凝聚相产物 总结 基于 ML 模拟方法获得燃烧过程中涂层内分子数量、势能、固体或气体产物的动态演化过程。涂层在燃烧过程中生成的金属/非金属化合物可以在 EPS 表面形成保护层,起到良好的凝聚相阻燃作用。涂层能够释放大量 CO2 和 H2O 等不可燃气体,且所产生的金属化合物对活性自由基具有较高的捕捉能力,表现出良好的气相阻燃作用。这项模拟研究为 EPS 泡沫的阻燃提供了一种新的方法,在建筑、工业和交通运输领域具有广阔的应用前景。 参考文献 Bin Li, Chuanshen Wang, Yifu Xiang, Wei Zhang, Bin Yu, Jinzhang Jia, Meihua Lian, A cost-effective strategy to construct highly effective flame-retardant coatings of modified epoxy resin/layered double hydroxide/zinc borate for polystyrene foam, Construction and Building Materials, Volume 478, 141324 (2025) https://doi.org/10.1016/j.conbuildmat.2025.141324