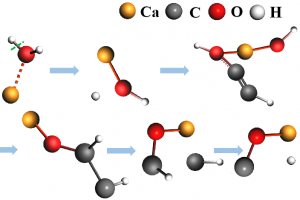
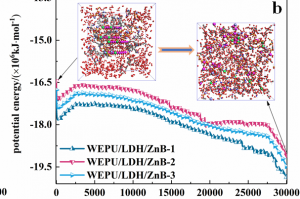
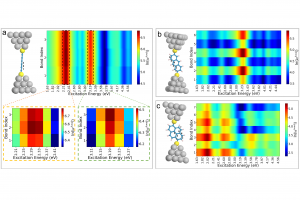
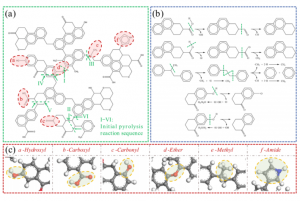
研究背景 全球能源转型亟需绿色制氢技术。生物质气化制氢虽具潜力,但水煤气变换反应(WGSR)受高浓度 CO₂ 逆向抑制,限制产氢效率。研究表明,添加 CaO 可通过原位吸附 CO₂ 显著提升 H₂ 产率(实验显示最高可倍增),同时催化焦油重整提升转化率 23–41%。然而,CaO 在复杂气化体系中的”吸附-催化”协同机制尚未明晰。通过联合 ReaxFF MD 与 DFT 模拟,探究 CaO 催化木质素气化制氢的反应机理,为设计高性能钙基催化剂提供理论依据。 研究内容 宁夏大学含碳基质气化课题组,采用 ReaxFF MD 与 DFT 结合的方法,揭示了 CaO 催化木质素气化制氢的多尺度协同机制。研究表明高温下 CaO 解离释放的高活性 Ca2+ 易与气化剂(H2O)或含氧有机物的氧部位结合,促进O-H、C-H、C-O 键断裂。此外,也探究了温度、水碳比(S/B)等因素对催化效果的影响。该研究结果有望为设计高性能钙基催化剂提供理论支撑。 图1. CaO 催化木质素气化反应路径模拟快照 CaO 在生物质气化过程中表现出明显的催化活性,其主要机理是通过降低反应势垒和加速化学键断裂显著提升气化效率。然而,在温度升高时,尽管 CaO 仍能保持一定程度的催化增强作用,但其功效会相应减弱。这种衰减可能与高温条件下催化活性位点的部分失活有关。因此,氧化钙催化剂的工作温度范围必须考虑催化性能和热稳定性之间的协同平衡,以便在工业气化情况下实现最佳催化效率。 图2. 不同温度下气化反应势能的演变过程 通过对不同体系气化产物的统计,发现 CaO 的催化效能呈现非线性温度响应,其通过降低特定反应能垒改变产物分布,但高温吸附饱和与热失活制约效率,需优化温度窗口以平衡反应路径与催化活性。 图3. 不同温度下的气化产物分子数(a)H2(b)CO(c)CH4(d)CO2 通过固定温度 3000 K,探讨了水蒸气与 CaO 在木质素气化中的协同作用,统计不同 S/B 比下 H2、CO、CO2 和 CH4 的分子个数。结果表明,H2 产量随 S/B 比增加而上升,但增速减缓;CO 产量在无 CaO 时增加,有 CaO 时略有上升;CO₂ 在有 CaO 时显著增加,CH₄ 变化不大。作者认为提高 S/B 促进水煤气反应,但高温高 S/B 下 CaO 吸附易饱和。 图4. 不同 S/B 条件下气化产物的分子个数(a)H2(b)CO(c)CH4(d)CO2 采用 AMS 软件的 ReaaxFF 及其 ChemTraYzer 2.0 基元反应分析功能,统计气化时发生的反应及其次数。明确了 CaO 通过调控电子结构(静电极化/能隙窄化)实现生物质高效气化,但高温导致的团聚与逆反应制约催化效率,需通过活性位点工程平衡稳定性与活性。 图5. 生物质气化过程中 CaO 催化反应机理 总结 本文利用 AMS 软件,探究了 CaO 催化生物质气化制氢的多尺度协同机制。研究发现,温度对催化活性呈非线性关系,高温促进气化剂解离,但导致 CaO 团聚失活,为优化操作窗口提供理论依据。另外,提高 […]