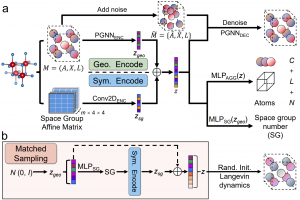
研究背景 自旋电子学通过利用电子自旋而不仅是电荷来存储和处理信息。当前磁随机存储器等器件大多依赖铁磁体的宏观磁矩进行读写,但铁磁体容易受到外部磁场干扰,并会产生杂散场,限制器件高密度集成。反铁磁体(AFMs)中相邻自旋反向排列,净磁矩为零,因此天然具备抗磁场干扰、低杂散场和太赫兹尺度超快磁动力学等优势。 然而,判定一个材料是否具有反铁磁基态,并非仅由化学组成决定。在高对称性的磁性晶体结构中,位于不同空间格点的磁性原子,其自旋构型可能更容易满足空间反演和时间反演的联合对称性。这种对称性将导致自旋向上与自旋向下的能带完全简并,从而形成反铁磁序。因此,反铁磁材料的发现本质上是一个多维度问题,需要同时兼顾化学空间的广度、几何结构的合理性以及晶体对称性等多重约束。 近年来,深度生成模型在晶体逆向设计中展现出巨大潜力1-4。但现有生成模型普遍缺乏对晶体对称性的显式编码,导致其生成高对称结构的效率偏低3,4,往往更倾向于产生低对称性乃至 P1 空间群的结构,难以系统地探索反铁磁材料更可能涌现的高对称空间。 研究内容 杭州电子科技大学张正明副教授等人提出了一种名为SG‑CDVAE的生成框架。他们将晶体对称性这一关键物理信息嵌入到生成模型架构中,从而实现对高对称反铁磁材料的定向设计。SG-CDVAE 以 CDVAE 为基础,同时编码晶体几何结构和空间群仿射矩阵:通过周期图神经网络编码器提取几何隐变量 zgeo,通过二维卷积编码器提取对称性隐变量 zsg,两者拼接后形成融合几何与对称信息的完整隐变量 z。在训练阶段,模型一方面用 z 预测组成、晶格和原子数,另一方面用 zgeo 预测空间群编号;扩散解码器则在 z 条件下对加入噪声的原子坐标和原子类型进行去噪,重构晶体结构。这样做的目标是让隐空间不只学到局部几何,还能学习全局对称性。在生成阶段,引入了匹配采样(matched sampling)策略。首先随机采样 zgeo,预测其对应的空间群及仿射矩阵,再编码得到匹配的 zsg,从而确保生成的晶体结构在初始时就具有较高的对称性。对比实验表明, SG‑CDVAE生成的高对称结构(空间群编号 SG ≥ 25)比例达 19.62%,远高于原始 CDVAE(3.85%)。 图1. SG-CDVAE 同时编码晶体几何与空间群仿射矩阵,并通过匹配采样生成高对称结构 图2. SG-CDVAE显著提升空间群SG >= 25 高对称结构生成比例,并给出不同晶系分布 为快速评估生成晶体的稳定性和磁性,构建了基于 CHGNet5 的高通量筛选流程。首先,用 SG-CDVAE 大量生成晶体结构;然后保留磁性原子数为偶数的结构,这是因为补偿反铁磁序通常需要成对的磁性原子格点;接着使用 CHGNet 快速弛豫晶格和原子位置,并保留形成能 FE < 0 eV/atom 的结构。磁性判断采用两级分类器区分磁有序/非磁,以及反铁磁/铁磁/亚铁磁。最终筛出 80 个 SG >= 25 的高对称反铁磁候选。 图3. 基于 MLIP 的高通量AFM筛选工作流程 […]