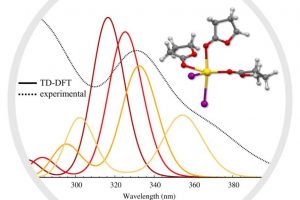
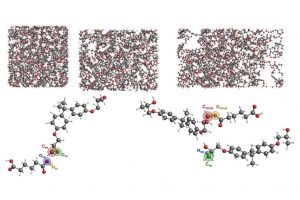
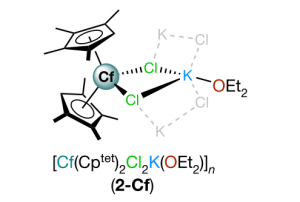
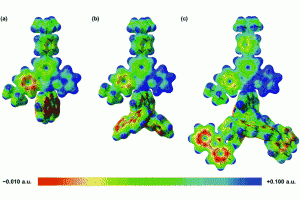
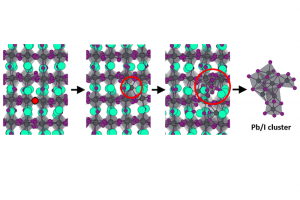
溶液合成是制备光电用金属卤化物钙钛矿最常用的方法之一。从溶液中控制钙钛矿的生长对于获得高质量的材料至关重要,这也需要研究者对钙钛矿前驱体化学有深入的了解。事实上,起始材料(盐、溶剂、添加剂)的选择决定了钙钛矿本身的最终形态和结晶度。因此,确定钙钛矿溶液制备过程中形成的溶剂化碘化物(即碘化铅络合物)的构成,对理解溶液与材料性质之间的关联非常重要。尤其是溶剂和添加剂如何影响碘化物平衡,这将使我们在理解这些材料的行为方面得到进一步的认知。 为了达到这个目标,佩鲁贾大学和CNR SCITEC的研究人员开发了一个综合的实验和计算框架,它包括UV/Vis吸收光谱和密度泛函理论(DFT)模拟。静态计算和分子动力学用于揭示溶剂化碘化物模型结构的特性,然后对其进行TDDFT计算,得到包含自旋轨道耦合(SOC)效应的精确其光学行为。 理论和实验吸收光谱之间的极好一致性,让研究人员发现可信的溶剂化钙钛矿物种与配位溶剂。在一系列论文中,报道了二甲亚砜(DMSO)、N,N-二甲基甲酰胺(DMF)[1]和γ-羟基丁酸内酯(GBL)[2]中的钙钛矿溶剂化结构。通过比较实验光谱和计算光谱,还研究了水[3]和PbCl2铅盐前驱体[4]对碘化物平衡的影响。这些研究揭示了钙钛矿前驱体溶液的不同组分如何影响溶剂化复合物的性质,这些组分对钙钛矿的生长方式、形态和最终材料中发现的缺陷类型有直接影响,从而影响其在太阳能电池中的性能。 输入文件下载:*.ams,*.run 参考文献: [1] Radicchi, E.; Mosconi, E.; Elisei, F.; Nunzi, F.; De Angelis, F. Understanding the Solution Chemistry of Lead Halide Perovskites Precursors. ACS Appl. Energy Mater. 2019, 2, 3400–3409. [2] Radicchi, E.; Kachmar, A.; Mosconi, E.; Bizzarri, B.; Nunzi, F.; De Angelis, F. Structural and Optical Properties of Solvated PbI2 in γ-Butyrolactone: Insight into […]