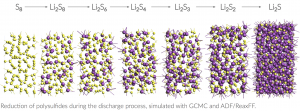
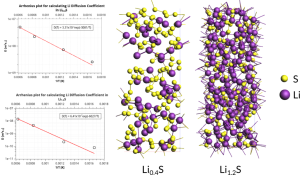
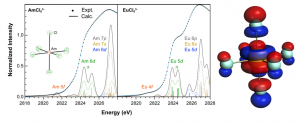
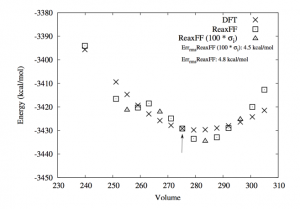
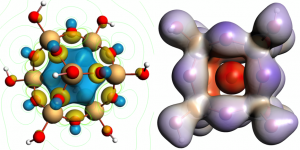
英文教程 点击此处下载英文教程 英文教程阅读说明 准备知识 该功能版本号≥2017.211,下载地址:https://www.scm.com/support/downloads/development-snapshots/,svnRevision大于r62841的都可以 熟悉ReaxFF建模、保存任务、提交任务等基本操作 熟悉巨正则系综的含义更好 知道如何在命令行里面运行命令(本教程需要在命令行运行一次分析命令,得到电压特性数据) 知道如何将数据从文本格式转移到Excel中,并生成曲线,参考:如何从图谱中导出数据到Excel表格 阅读说明 该教程非常详细,实际上初学者也可以照着说明一步一步做出来,当然结果会有差异,这是正常的。本教程主要包含如下几个小节: 从外部导入Alpha硫晶体的*.cif文件,分别采用NPT系综低温弛豫(ADFinput > ReaxFF > Main > Task: Molecular Dynamics)、能量最小化(ADFinput > ReaxFF > Main > Task: Geometry Optimization)来得到其平衡结构。需要说明的是,教程中能量最小化得到的能量是-8535.99 kcal/mol,实际我们得到的能量可能更低。 计算Li原子的化学势:计算体心立方锂晶体的8*8*8超胞(共512个Li原子)的能量,除以512,即得到Li原子的化学势-37.70kcal/mol 对Alpha硫晶体进行巨正则系综蒙特卡洛模拟 从上一步GCMC模拟生成的*.rxkf文件中,抽取电压特性等数据:将LiVoltageProfile.py文件和*.rxkf放到同一个文件夹内,在ADF环境变量对当前命令行生效的前提下,在命令行执行:startpython LiVoltageProfile.py gcmc_test.rxkf。将生成一个文件,名为voltage_profile.out。该文件实际上是一个文本文件,给出了4列数据,其中第二列是LixS中的x值,第三列是电压,第四列是体积变化。用户可以自行用这两列数据生成教程中的曲线 上面这几步的计算模拟文件(请点击此处下载),在阅读困难的情况下,可以辅助您阅读教程,但由于GCMC这一步很耗时间(对应文件gcmc_test.adf),可能需要一天左右才能完成,因此设置的参数数值放宽了条件,没有教程里面那么严格,因此这一步仅供参考。