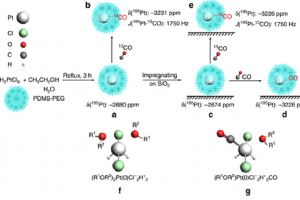
文献资料:Kairui Liu, Guangjin Hou, Jingbo Mao, Zhanwei Xu, Peifang Yan, Huixiang Li, Xinwen Guo, Shi Bai & Z. Conrad Zhang, Genesis of electron deficient Pt1(0) in PDMS-PEG aggregates, Nature Communications volume 10, Article number: 996 (2019) 虽然载体表面稳定单原子的报道已经屡见不鲜,但在液体介质中合成原位还原的稳定弱配位分散金属原子更具挑战性。本文报道了在液体PDMS-PEG中还原H2PtCl6,形成单核缺电子Pt1(0) (Pt1@PDMS-PEG),该还原反应通过紫外-可见光谱、远红外光谱和X-射线光电子能谱得到证实。 CO红外光谱、195Pt与13C核磁共振谱证实Pt1(0)以近似八面体结构(R1OR2)2Pt(0)Cl2H2存在,其中R1、R2 分别是H、C或Si基团。通过比较实验与理论计算的195Pt核磁共振谱,验证了弱配位结构(R1OR2)2Pt(0)Cl2H2以及缺电子Pt1(0) 。质子态H+与Cl−形成类似HCl的弱配位,用碱中和生成铂纳米粒子。Pt1@PDMS-PEG在石蜡氢化硅烷化中表现出超高的活性以及最终加成物选择性。 NMR位移计算使用ADF模块,利用ZORA方法考虑重元素Pt的相对论效应。