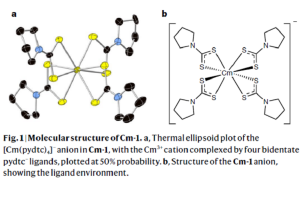
锔在锕系元素中是独一无二的,因为它的半填充5f7壳层比其他5fn结构的能量更低,因此既难于氧化还原,(5f壳层)又不易形成化学键。这一点对钆(钆是镧系中锔的类似物)更为明显,因为相对于锔的5f轨道,钆的4f轨道更为紧缩。 然而在高压下,锔的5f电子从局域化状态转变为巡游态。这种转变形成一种晶体结构,这种结构由锔原子之间的磁相互作用决定。那么是否也可以通过施加压力来改变锔(III)-配体相互作用中的前沿金属轨道,从而诱导形成具有一定共价性的金属-配体键? 弗洛里达州立大学Thomas E. Albrecht-Schönzart,纽约州立大学Eva Zurek、Jochen Autschbach,亚琛工业大学Manfred Speldrich等课题组合作,报道了在高压(高达11GPa)下,[Cm(pydtc)4]–(pydtc,吡咯烷二硫代氨基甲酸基)的锔-硫键中,5f/6d轨道角色变化的实验与计算结果。对键性质的计算与NLMO分析,采用AMS软件ADF模块完成。计算结果表明,锔的5f轨道对锔-硫键的贡献在高压下显著增强,在11GPa时翻倍。 与[Cm(pydtc)4]–光谱中观察到的变化相比,加压后[Nd(pydtc)4]–的吸收光谱中f-f 跃迁,以及Cm(III)苯六甲酸盐的f–f 跃迁发射光谱的变化较小,这是由于它们的键性质受压力的扰动较小。 这表明,锕系化合物的共价性,即使对同一离子也是复杂的,但研究压力对锕系化合物的影响,可以指导配体的选择。 参考文献: Joseph M. Sperling, Evan J. Warzecha, Cristian Celis-Barros, Dumitru-Claudiu Sergentu, Xiaoyu Wang, Bonnie E. Klamm, Cory J. Windorff, Alyssa N. Gaiser, Frankie D. White, Drake A. Beery, Alexander T. Chemey, Megan A. Whitefoot, Brian N. Long, Kenneth Hanson, Paul Kögerler, Manfred Speldrich, Eva Zurek, […]