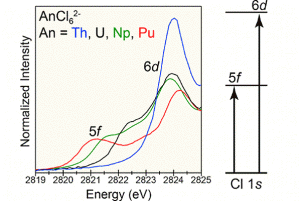
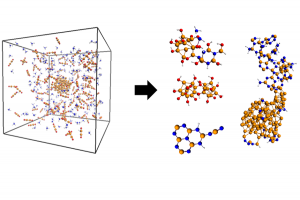
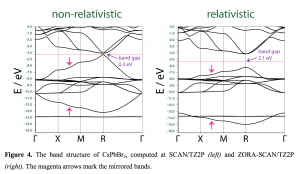
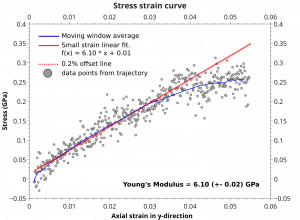
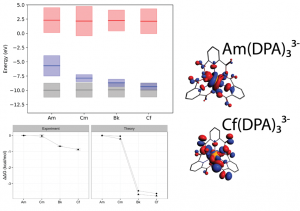
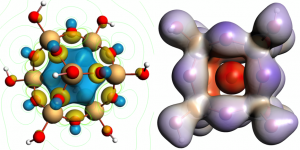
文献资料:Jing Su, Enrique R. Batista, Kevin S. Boland, Sharon E. Bone, Joseph A. Bradley, Samantha K. Cary, David L. Clark, Steven D. Conradson, Alex S. Ditter, Nikolas Kaltsoyannis, Jason M. Keith, Andrew Kerridge, Stosh A. Kozimor, Matthias W. Löble, Richard L. Martin, Stefan G. Minasian, Veronika Mocko, Henry S. La Pierre, Gerald T. Seidler, David […]