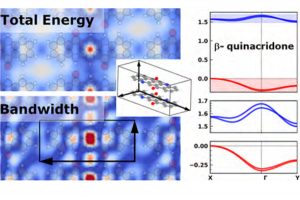
分子单体之间的电子耦合是决定有机半导体电荷输运的一个关键因素,而这直接受分子排布方式所影响。 Christian Winkler等研究了喹吖啶酮不同晶相下的分子间的相互作用,及其对能量稳定性的影响。为了深入研究这种影响,作者从α晶相喹吖啶酮为原型创建了共面的模型晶体,从而系统地比较电子耦合、总能量与位移的相关性。 通过这种方法确认,泡利排斥以及轨道的再次杂化,促使该体系倾向于电子耦合最低的晶体结构。这种趋势具有一定普适性,并五苯类似物也有这种趋势。 这表明,高性能的材料设计不能依赖有机半导体π共轭骨架结构“自然地”组装,必须引入官能团,引导晶体向更有利的结构方向发展。其中,以短轴位移为目标或实现相当大的长轴位移的策略,值得深入研究。 本文使用了AMS中的BAND模块的pEDA功能,将分子间的相互作用能分解为泡利排斥能、轨道相互作用能、静电作用能。并使用ZORA方法考虑相对论效应对电子动能的影响。 参考文献: Christian Winkler, Andreas Jeindl, Florian Mayer, Oliver T. Hofmann, Ralf Tonner, and Egbert Zojer, Understanding the Correlation between Electronic Coupling and Energetic Stability of Molecular Crystal Polymorphs: The Instructive Case of Quinacridone, Chem. Mater. 2019, 31, 7054–7069