
背景
二维量子材料有望改变传统电子学,实现涵盖化学科学的广泛应用。为了研究单层(1L)或多层过渡金属二卤化物(TMD)中的热输运,作者探索了密度泛函理论(DFT)和训练算法的结合,生成了模拟 1L-MoS2、1L-WS2 及其合金(混合结构)的矩张量势(MTP)力场,并展示了理论技术的协同将在该领域发挥重要作用。从高性能计算的角度来看,所产生的非常方便的原子间势(或分子间势),可用于预测量子材料对热扰动或其他驱动力的响应。
研究内容
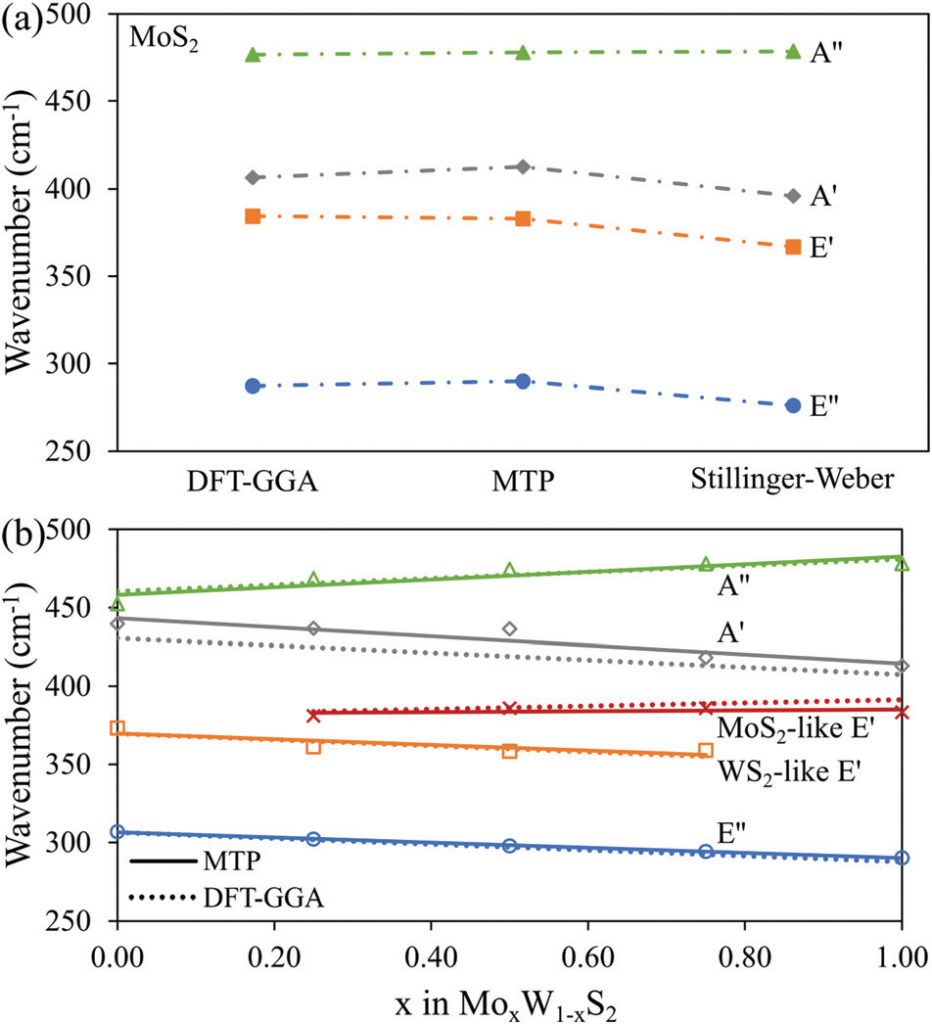
作者将数百个来自原子位置微扰和分子动力学轨迹的单层结构作为训练集用于 MTP 力场的训练,对比了用所得到的 MTP 力场计算的声子性质与 DFT 和 SW 力场计算的结果。训练得到的 MTP 力场很好的描述了体系的振动特性及其热导率。
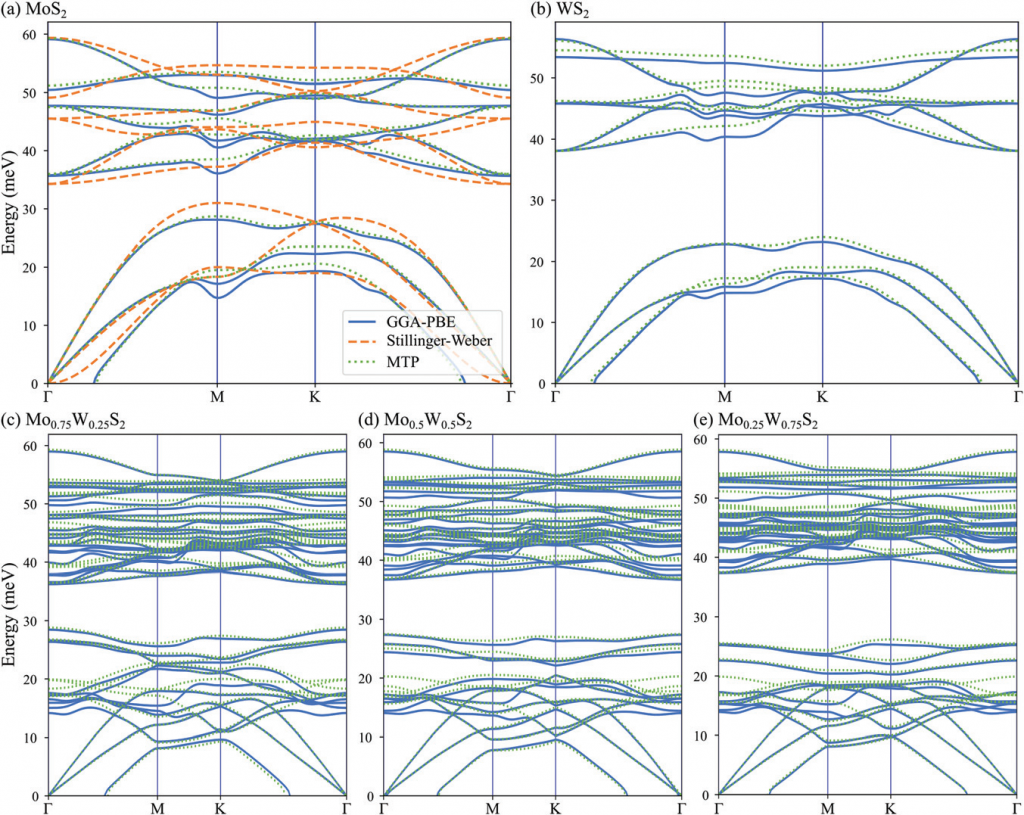
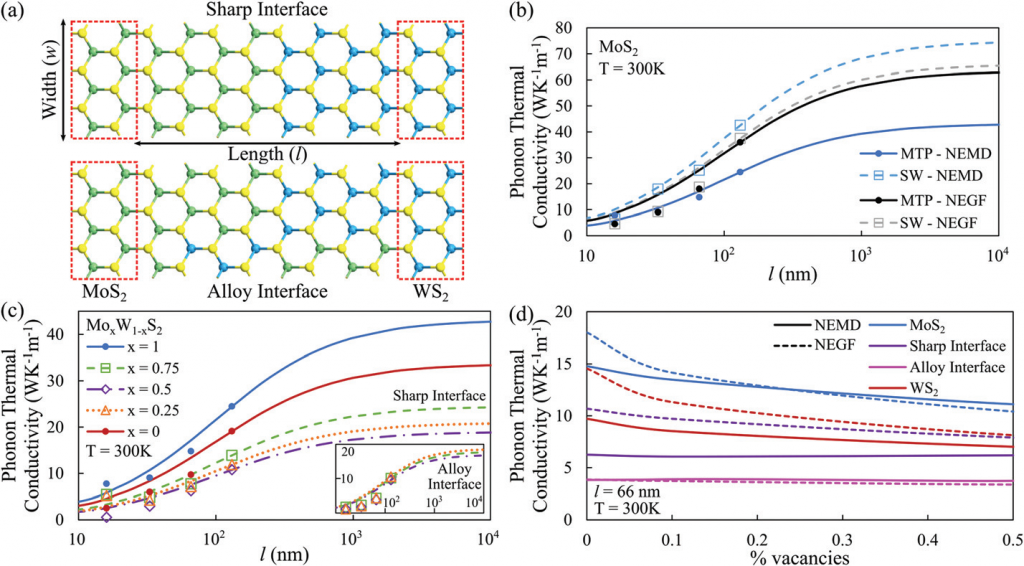
作者将得到的 MTP 力场用于晶格声子热导率的计算,对比了非平衡分子动力学(NEMD)和非平衡格林函数(NEGF)两种方法的结果。
结论
作者的结果表明,机器学习的 MTP 力场可以准确地再现原始和合金 1L TMD 的声子结构和热传输特性。1L-MoxW1-xS2 合金结构在不同浓度下具有独特的声子谱特征,但观察到的热传输特性对组分的依赖性非常低,这使得通过混合不同原子种类来设计类似材料的热传输特性成为可能。此外,2D 合金结构对硫空位不敏感,热导率几乎不受硫空位的影响,这一行为可能有助于微调材料的热性能,以用于热管理和能量存储和转换应用。这也提示我们可以不依赖于获得无缺陷晶体材料的昂贵技术来制造器件。最后,模拟精度在更大程度上取决于参考 DFT 方法和训练集,因此,对于实际模拟体系来说,可以不需要计算成本高、冗长的优化步骤,就能获得合理的结果。
参考
- Juan M. Marmolejo-Tejada et al. Thermal properties of single-layer MoS2–WS2 alloys enabled by machine-learned interatomic potentials. Chem. Commun., 2022, 58, 6902–6905. https://doi.org/10.1039/D2CC02519A
- 更多信息: