
研究背景
传统的成键规则塑造了对分子结构的理解,但平面超配位原子(如平面四配位碳 ptC)的出现打破了这一范式。随着研究从四配位向更高配位数拓展,基于电子离域和配体环状排列的设计策略已成功应用于碳、氮及部分金属主族元素,实现了平面五配位乃至六配位物种的稳定。然而,卤素因具有极高电负性,长期被视为平面超配位的“禁区”,近期仅有的平面四配位氟(ptF)研究也多涉及准平面结构。近日,吉林大学崔中华教授团队在 Angew. Chem. Int. Ed. 上发表了重要研究,通过严谨的计算化学预测,首次在 Li5X6−(X=F, Cl, Br)团簇中确立了具有 D5h 对称性的平面五配位卤素(ppX)全局极小值结构。这一发现不仅拓宽了平面超配位的版图,更揭示了在高电负性原子上实现非常规配位的独特策略。
研究内容
基于卤素离子(X⁻)区别于中性卤原子的特性:其全充满的价电子层不仅可作为弱电子供体,更能作为强静电相互作用源,因此将X⁻束缚于由锂原子构成的高极性(Li⁺X⁻)ₙ环中,利用中心卤素与锂之间的强静电相互作用有望实现ppX结构。
初步几何探索表明,X©Li₄X₄⁻和X©Li₆X₆⁻(图1)因环尺寸与中心原子不匹配存在较大虚频,而X©Li₅X₅⁻(X=F, Cl, Br)(图1)在PBE0-D3、TPSS-D3和M06-2X-D3等多种理论水平下均被证实为势能面上的真实极小值。随后,通过目标导向遗传算法结合多级计算策略(从def2-SVP基组优化到aug-cc-pVTZ基组重优化,再到CCSD(T)单点能计算及零点能校正),对体系进行了全局异构体搜索(图2)。结果显示所有异构体的T1诊断值均小于0.02,证实了单参考计算方法的可靠性,并确立了ppX结构的稳定地位。
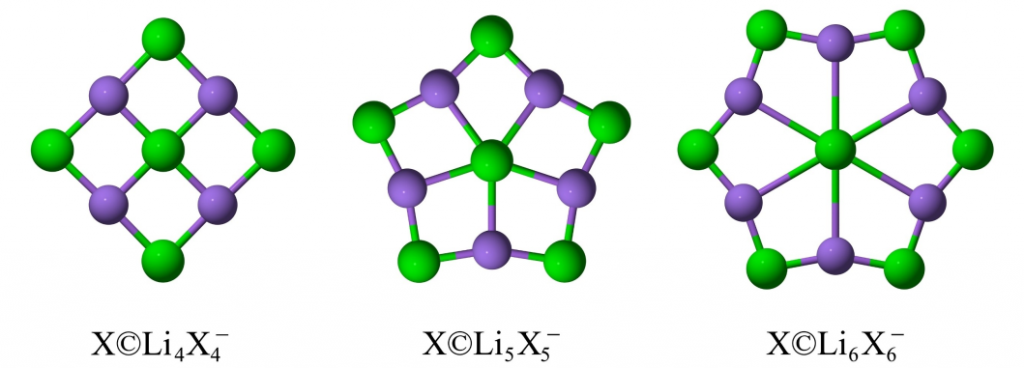
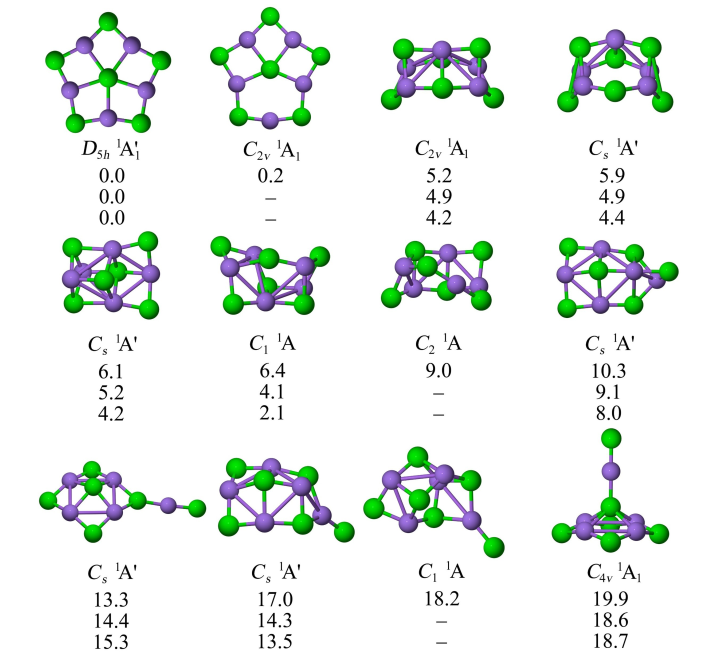
图 2 展示了 Li5X6−(X=F, Cl, Br)的部分低能异构体。在所有情况下,能量最低的结构均为 ppX 构型,具有 1A1′ 电子态和 D5h 对称性。其中 ppF 与 ptF 结构能量相近且存在快速互变,而 Cl 和 Br 体系的 ptX 结构则不稳定,收敛至 ppX。该类化合物的结构显著优于其他三维异构体。
2 在
CCSD(T)/aug-cc-pVTZ//PBE0-D3/aug-cc-pVTZ 水平下计算(含PBE0-D3/aug-cc-pVTZ零点能校正)得到的Li₅X₆ (X=F, Cl, Br) 低能异构体的结构及相对能量(单位:kcal/mol)
ppF、ppCl 和 ppBr的电子结构分析显示,中心 Li−Xin 键长(2.08、2.51 和 2.66 Å)略长于典型单键,Mayer 键级(0.15–0.23)揭示其在静电主导下仍具有不可忽视的共价特征。相比之下,外围 Li−Xout 键键长较短(1.72、2.21 和 2.38 Å)且键级较大(0.54–0.58)。而中心原子与外围原子间(Xin−Xout)距离较远(2.90、3.68 和 3.95 Å)且键级接近零,表明两者间无显著的相互作用。同样,较小的 MBO 值和 2.44–3.14 Å 的较大距离表明 ppX 中的 Li−Li 相互作用可以忽略不计(图3 (a) )。
自然电荷分析表明,ppX 原子与桥连卤素均带负电荷(−0.85 至 −0.92 |e|),而锂原子带正电荷(0.82–0.91 |e|),证实体系可表述为 X⁻©Li₅X₅,其稳定性主要源于 ppX 与 Li⁺ 间的强静电吸引,且高达 7.0 eV 以上的 HOMO-LUMO 能隙显示出极高的化学稳定性。AdNDP 分析揭示了两种互补的成键图像(图3(b)):中心 X⁻ 可视为拥有三对孤对电子(纯粹的离子键),或参与形成三个 6c–2e 离域键(暗示微弱电子离域)。
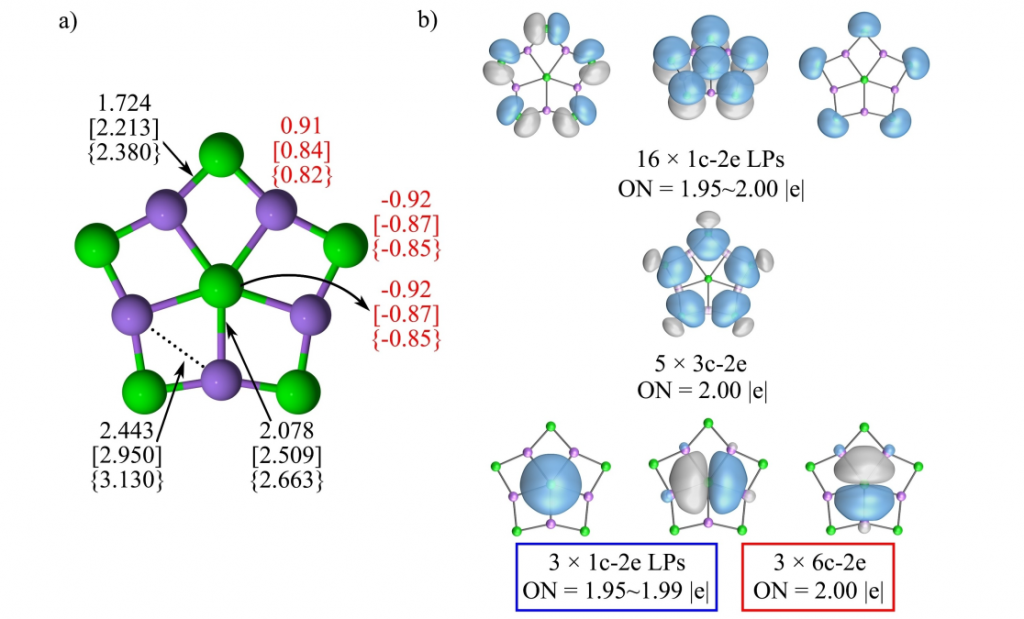
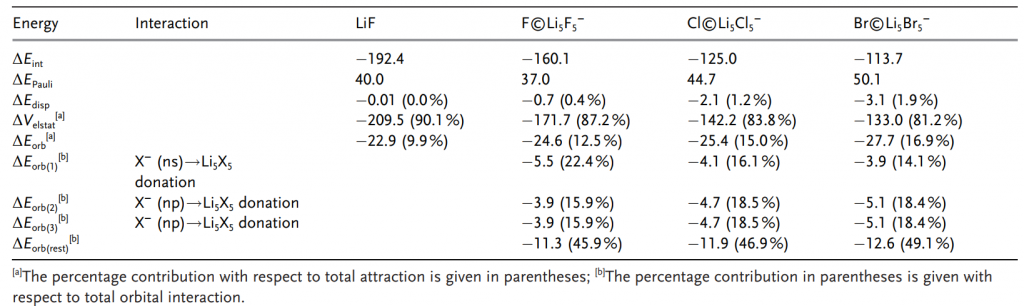
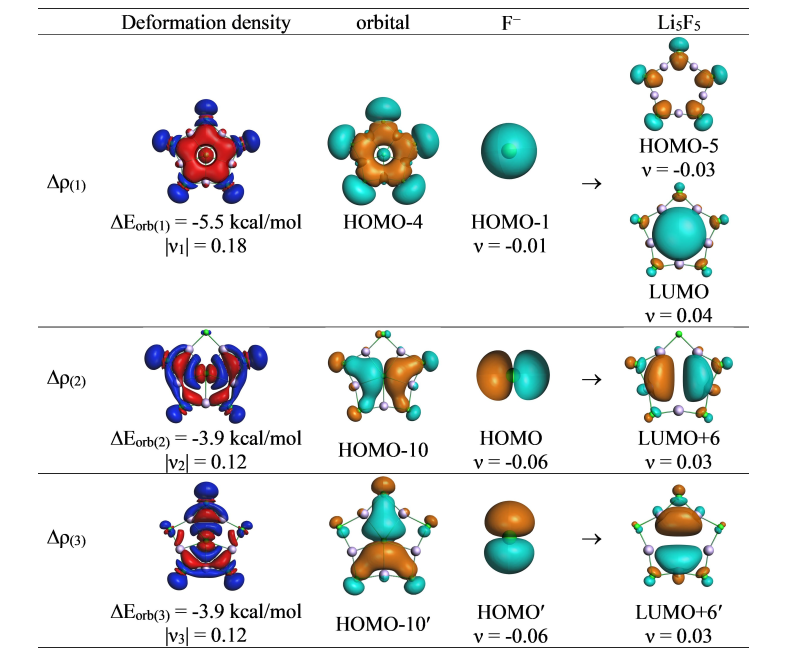
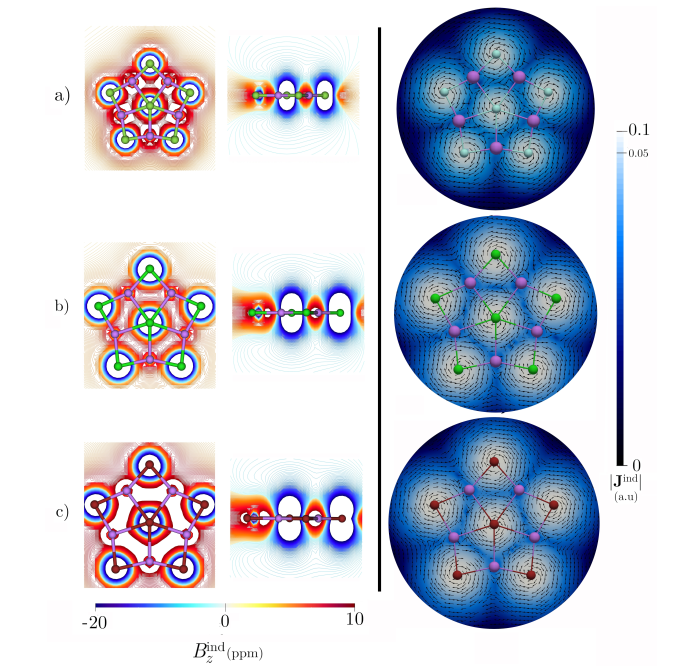
进一步基于 AMS 软件的 EDA-NOCV 分析,量化了成键本质:中心原子与其他部分之间,静电相互作用贡献了 81–87% (表1)的吸引能,占主导地位;轨道相互作用(ΔEorb)虽仅占 12–17%(表1),源于一个弱的 X−(ns)→Li₅X₅ 配位键和两个简并的 X−(np)→Li₅X₅ 配位键(图4)。与 LiF 分子的对比进一步确认了该体系的离子化合物属性。磁响应计算显示(图5),由于轨道参与度较低,体系缺乏全局环电流,环电流强度极低(<1 nA/T),表明其稳定性机制不同于传统依赖电子离域的平面超配位物种,而是由强静电相互作用主导。
表 1 在考虑离子碎片(Li⁺ (¹S) + F⁻ (¹S))时的 LiF(r=1.570 Å),以及考虑 Xin⁻ (S, ns²np⁶) 和 Li₅X₅ (S) 为相互作用碎片时的 X©Li₅X₅⁻(X=F, Cl, Br)的 EDA 结果。
ppX (X©Li₅X₅⁻) 在 300 K 下的 Born-Oppenheimer 分子动力学模拟结果显示,在约 100 ps 的模拟时间内,体系始终保持结构完整性和平面性,未出现异构化或结构转变迹象,面外位移的势能曲线表明平面结构相比三维构型具有显著的能量优势。同时,针对多种解离路径的热力学计算表明,所有解离能均为较高的正值(33–820 kcal/mol),其中最低能解离路径 Li₅X₆⁻ → LiX + Li₄X₅⁻ 也需克服 33–54 kcal/mol 的能垒。这些结果共同证实了 ppX 体系优异的动力学和热力学稳定性。
总结
本研究突破了卤素因高电负性而难以形成平面超配位的传统认知,在 Li₅X₆⁻ 团簇中成功鉴定出具有 D₅h 对称性的平面五配位卤素全局极小值结构。中心卤素以卤素离子形式嵌入平面 Li₅X₅ 环,主要依靠强静电相互作用及非零的共价成分稳定,虽存在局部环电流但缺乏全局电子离域。该类体系展现出优异的动力学与热力学稳定性,有望通过气相阴离子光电子能谱观测,为深化化学键理论及设计非传统分子架构提供了重要参考。
参考文献
- Cui L, Miao L, Orozco‐Ic M, et al. Planar pentacoordinate halogens[J]. Angewandte Chemie, 2025, 137(4): e202416057. https://doi.org/10.1002/anie.202416057