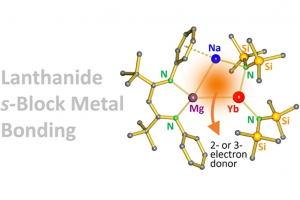
研究背景 镧系金属由于其 4f 轨道高度屏蔽、径向扩展性弱,难以与其他原子轨道有效重叠,长期被视为不参与共价成键的“惰性”金属。在多数配合物中,镧系元素仅通过静电作用与配体中的 O 或 N 等硬碱相互作用,呈现典型的离子型键合行为。 相比之下,镧系–金属之间的直接金属–金属键(Ln–M)极为罕见,已知例子多限于 Ln–p 区金属(如 Sn、Ga、Al)体系,且多依赖阴离子桥连或范德华相互作用。镧系–s区金属(如Mg、Ca)之间的共价成键此前从未被实验证实,主要因两者都电正性极强、缺乏成键轨道匹配。 研究内容 为探索镧系与 s 区金属之间是否存在可控、可设计的 M–M 成键行为,德国埃尔朗根-纽伦堡大学 Harder 课题组构建了一个以中性镁配体为桥、实现 Mg–Yb 直接成键的复合物。该工作于 2025 年发表于 JACS,并以 Mg–Ca 体系为参照,首次实现 s 区–镧系金属间的系统对比研究。 图1. Mg–Yb直接成键的复合物结构 作者使用 [(BDI*)MgNa]₂ 作为反应平台,该中性二聚体含有两个低价镁中心(形式上为 Mg⁰),通过 Na⁺ 桥联。其与 Yb[N(SiMe₃)₂]₂ 反应,得到目标化合物 2-Yb,其晶体结构清晰显示出 Mg–Yb 之间存在明确的成键距离(3.466 Å),且整体框架与已报道的 2-Ca 结构几乎完全一致。 图2. (a) 2 -Yb、拟合 2 -Yb(红色)和 2 -Ca(绿色)(b)5 -Yb、(c) 分解产物 6 的晶体结构 图3. 含 […]