
BAND 概述
BAND用于周期性边界条件体系的第一性原理计算,因此主要用于三维晶体、二维固体表面与一维纳米线、纳米管、聚合物体系。能够对材料的谱学性质、光的吸收与折射、静电势、电子密度、原子电荷、费米面、有效质量、态密度、能带与化学键分析,电荷分析与谱学性质,表面化学反应、表面吸附问题进行研究。与流行平面波软件相比,BAND侧重材料化学计算,例如等。
BAND 的优势
- 效率:三维小体系计算效率较低,三维大体系、二维、一维周期性体系的计算效率比平面波方法高,杂化泛函的计算效率比平面波方方法高的多。
- MOF、COF等材料:BAND的基组是STO/NO,而不是平面波,因此真空区域不会显著增加计算量,因此对于Cell体积庞大,真空比例很高的MOF、COF等材料,计算效率远高于平面波方法
- 超重元素的精确计算:BAND不依赖赝势,而是精确的全电子基组
- 擅长成键分析:
- 键能分解分析pEDA:将键能拆分为静电作用、泡利排斥、轨道相互作用(即成键时,电子转移带来的能量降低量)
- 基于pEDA的NOCV:详细展示成键过程中,电子在不同晶体轨道中的转移情况,例如表面吸附时,小分子与晶体表面之间的电子转移情况
- 从化学键角度分析能带的COOP方法,优于COHP方法
- 化学相关功能非常丰富、易用:
- 过渡态搜索,采用NEB结合精确优化,大大提高过渡态计算的效率和精度
- PES Exploration:表面吸附位点、表面化学反应机理自动探索
- AIMD包含多种加速反应的方法
- 巨正则系综蒙特卡洛模拟
- 支持Win、Linux、MacOS系统
BAND 主要功能列表
- 周期性边界条件:
- 三维(体材料)
- 二维(材料表面)
- 一维(聚合物、纳米管线等)
- 0维(分子)周期性体系
- 相对论与重元素:
- 标量相对论
- 自旋轨道耦合
- 全电子基组、冻心基组
- 支持元素周期表所有元素,不依赖赝势
- 泛函:
- 各种常见GGA与meta-GGA泛函,如r2SCAN、极高效准确带隙的TASKxc、TASKCC等
- 适用于吸附与氢键问题的色散修正泛函,如:-D3(BJ)、-D4(EEQ)
- 杂化泛函HSE03、HSE06
- 丰富的libxc泛函库
- DFT-1/2方法得到更好的带隙、Hubbard U
- 基组:NTO/STO、纯STO基组、GTO基组
- 外场与特殊模型:
- 物理性质:
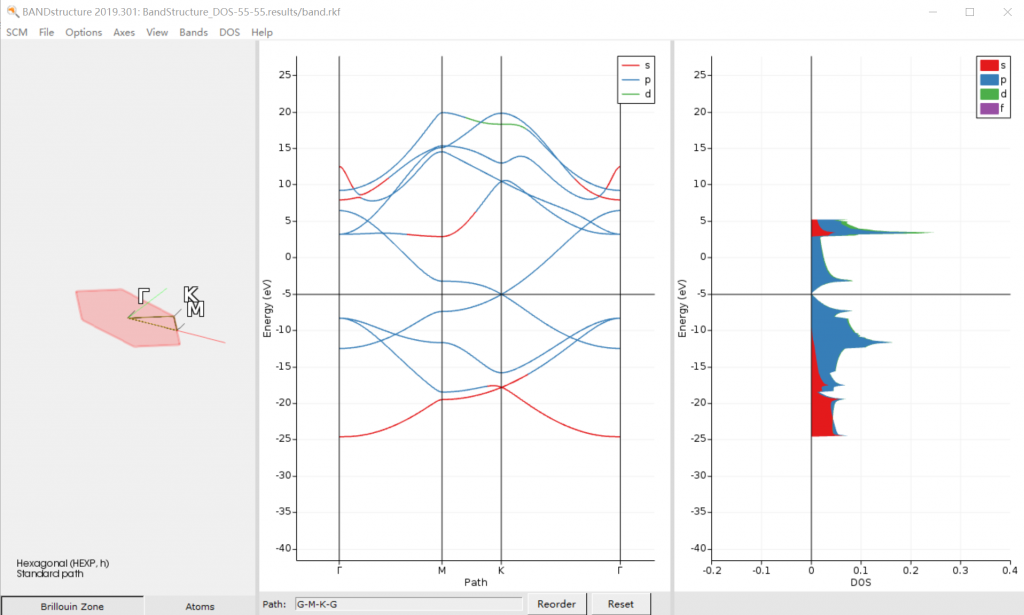

- 谱学性质:
- 磁化率、极化率、介电函数与光吸收、折射
- 单电子激发(教程)
- EELS、EFG、Q-tensor、 ESR、g-tensor、A-tensor
- 化学分析:
- AIM、原子电荷(Hirshfeld、Voronoi、CM5、Mulliken)
- 核电子密度、通过Laplacian电子密度与键关键点区分化学键类型
- Crystal Orbital Overlap Populations (COOP)
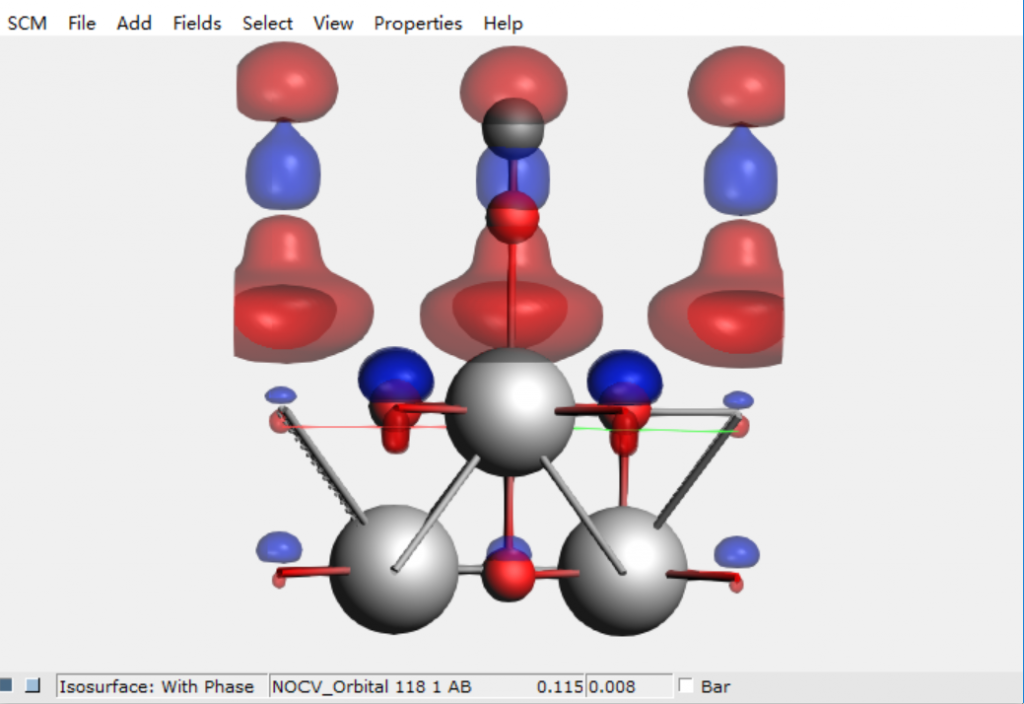
- 化学键分析:PEDA-NOCV键能分解与化学价自然轨道:
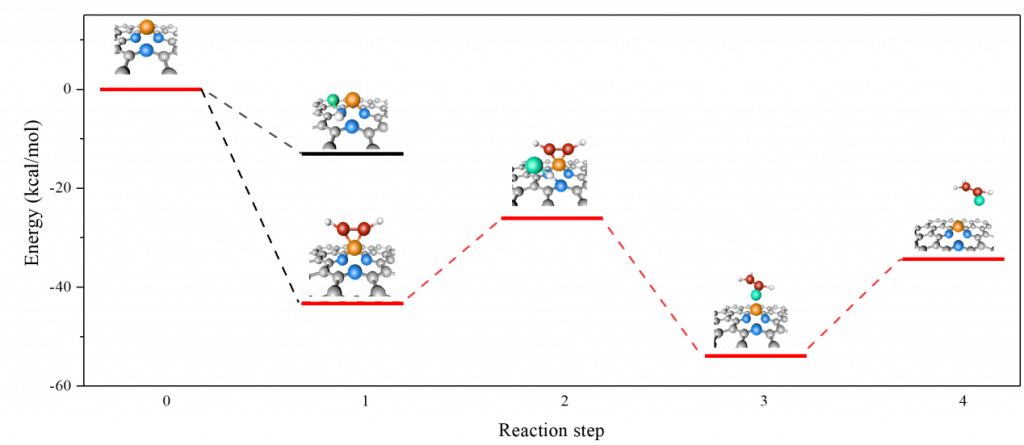
- 化学反应:
- 势能面扫描(PES)、过渡态搜索 、能垒计算、表面吸附位点自动探索、表面化学反应机理自动探索
- AIMD、基元反应分析、化学反应速率常数计算
- 巨正则系综蒙特卡洛模拟
- 偶极较正:平面波方法只能通过添加真空层,来模仿低维度体系,但静电作用是长程作用,乃至50埃以外仍然有明显影响,一般通过添加真空层,并不能很好解决这个问题,因此平面波方法还会通过添加一个偶极较正,来近似地达到“屏蔽”层与层之间远程静电作用的效果,但这种屏蔽实际上也只能部分解决该问题。BAND不存在该问题,因此无需进行偶极较正。
Quantum ESPRESSO 概述
AMS2024版将 Quantum ESPRESSO version 7.1 完全集成,功能与、赝势均进行了升级,并支持用Quantum ESPRESSO生成训练集,调用AdvancedWF模块的ParAMS、MD Active Learning功能,训练、优化机器学习势、ReaxFF力场。
Quantum ESPRESSO基于密度泛函理论、平面波和赝势方法,用于电子结构计算和纳米级材料建模,与BAND在效率与功能上具有互补之处。主要功能包括:
- AIMD 第一性原理分子动力学
- Berry相
- Hubbard U、外加电场(匀强电场、锯齿电场)
- 旋轨耦合、色散修正(-D2、-D3、-TS、-MBD、-XDM)
- 结构优化
- 晶格常数优化
- 能带、态密度计算
- 铁磁、反铁磁体系能带、态密度、pDOS计算、设置初始自旋/磁矩
- 表面化学反应计算
- 静电势分布、ELF、RDG、RDG-color
- 材料应力与原子受力
- 表面吸附位点自动探索
- 表面化学反应机理自动探索
AMS 中 Quantum ESPRESSO的优势
- 完全集成到 AMS 中,因此 AMS 驱动中的很多方法,都可以调用Quantum ESPRESSO
- 巨正则系综蒙特卡洛模拟
- AIMD
- 过渡态搜索,能够结合 NEB 与精确优化,提高效率
- PES Exploration:表面吸附位点、表面化学反应机理自动探索
- 支持生成 ReaxFF 训练集
案例与教程
- 进入AMS知识库,阅读BAND、Quantum ESPRESSO最新应用案例
- BAND、Quantum ESPRESSO中文教程