
研究背景
电催化 CO2 还原反应(CO2RR)能够在较温和条件下将 CO2 转化为高附加值化学品,是当前碳资源转化与能源化学领域的重要研究方向。其中,乙烯、乙醇等 C2 产物不仅附加值更高,也更能体现催化剂在 C–C 耦合步骤上的调控能力。相比 C1 产物,C2 产物的生成涉及更多中间体和更复杂的反应分叉,因此,如何从原子尺度揭示催化剂结构、反应路径与产物选择性之间的关系,一直是 CO2RR 理论研究中的关键问题。
双原子催化剂由于具有相邻双活性中心和可调电子结构,在促进 CO2 活化、关键中间体稳定以及 C–C 耦合方面展现出独特优势,近年来受到广泛关注。围绕这一方向,相关研究构建了 Co 基双原子氮掺杂石墨烯模型,系统比较不同双原子组合和不同位点构型下的稳定性、反应活性与产物选择性,为高选择性 CO2RR 催化剂设计提供理论依据。
在这类研究中,计算平台的适配性同样十分重要。AMS 软件中的 BAND 模块能够直接处理二维周期体系,适用于石墨烯、氮掺杂碳材料及表面催化模型;同时,结构优化、吸附能计算、溶剂模型、自由能分析以及电荷和轨道相互作用分析可以在同一平台内完成,因此非常适合开展这类涉及构型、吸附、路径、机理等全流程的理论研究。
研究内容
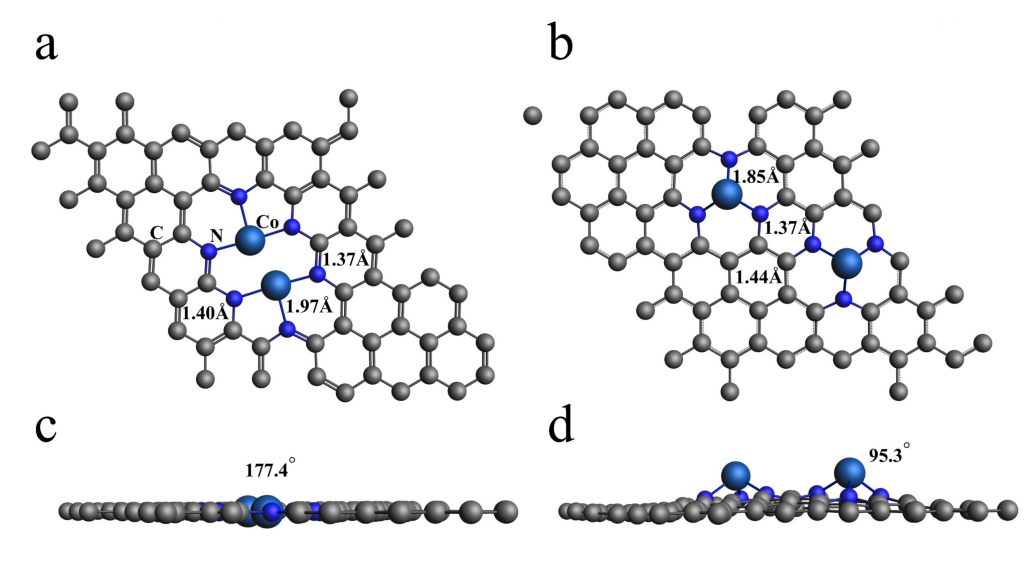
云南大学刘世熙课题组采用 AMS 计算软件中的 BAND 模块,设计了两种构型的 DACs 首先计算了不同构型载体的形成能以及其对金属原子的吸附能,结果表明a构型载体具有更低的形成能,同时对过渡金属原子的吸附能力也更强。此外,通过 Bader 电荷分析、PDOS 及 COOP 计算,进一步验证催化剂中 M1 和 M2 的 3d 轨道与 N 的 2p 轨道之间存在明显的成键作用,从而揭示了催化剂的稳定性。
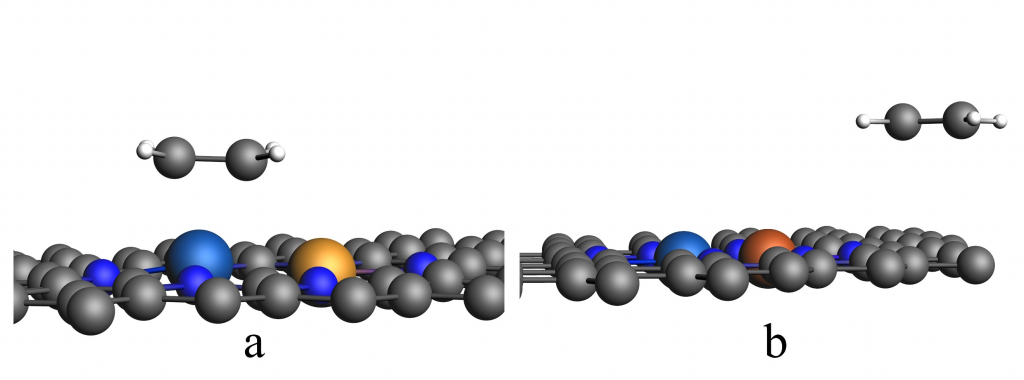
采用 BAND 模块,系统计算了 CO2、H、COOH、OCHO 等分子及中间体在 M1N3-M2N3@NGr 催化剂表面的吸附行为,确定了各反应中间体在催化剂上最稳定的吸附构型。其中 *CO2 的吸附计算结果显示,在
M1M2a催化剂上*CO2具有更小的夹角(135°~143.9°),同时, pEDA 与电荷分析揭示,M1M2a 不仅展现出更优异的 CO2 活化能力,同时也具备更出色的 CO2RR 催化性能。
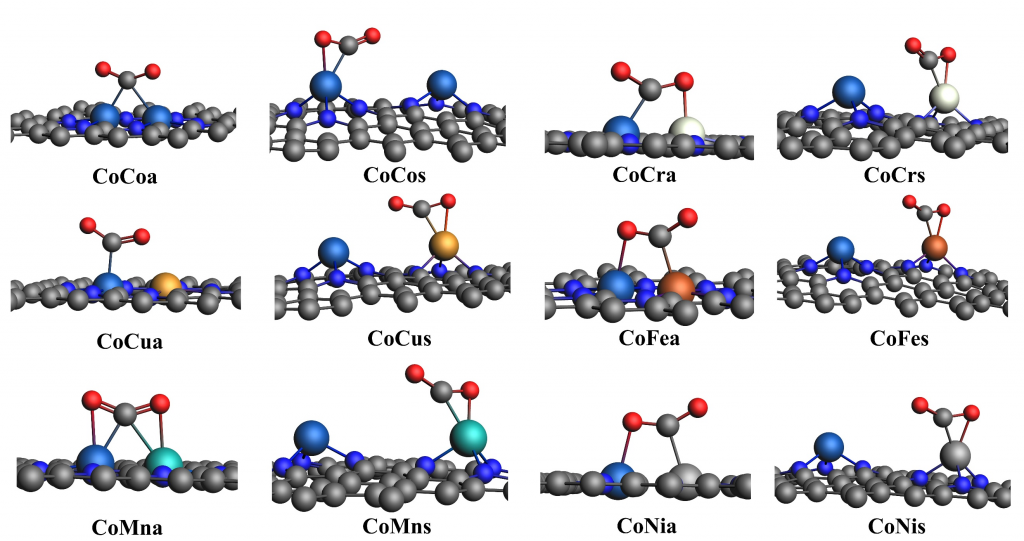
为了评估催化剂的性能与选择性,本研究计算了各步基元反应的吉布斯自由能。首先,通过比较 ΔG*H、ΔG*COOH 和 ΔG*OCHO 这三个值来排除析氢反应(HER)的影响,结果显示 12 种催化剂均更倾向于发生 CO2RR。在此基础上,我们采用以下 3 个标准来判断催化剂对 C2 的选择性:(1) ΔG*COOH < ΔG*OCHO; (2) ΔG*CO < ΔG*OCHOH; (3) ΔG*CO*CO < ΔG*CHO (或 ΔG*CHO < ΔG*CO*CO 并且 ΔG*CHO*CO < ΔG*CHOH)。分析结果表明,a 构型催化剂均具有良好的 C2 选择性;而对于 s 构型,只有 CoCrs 以及 CoFes 催化剂满足以上三个标准,但是这两种催化剂后续氢化步骤自由能垒过高(分别为 1.86 eV 和 0.95 eV),从而导致其 CO2RR 的活性并不高。
通过对各步吉布斯自由能的分析,我们确定了 6 种 a 构型催化剂生成 C2 产物的途径。CoCoa、CoCra 和 CoNia 催化剂更倾向于生成乙醇,其中 CoNia 表现出最优的乙醇催化活性,其极限电位(UL)为 −0.68 V;CoCua 则更利于乙烷的生成 UL 为 −0.61 V);而 CoFea 和 CoMna 更倾向于生成乙烯,其中 CoFea 的乙烯催化活性最高,UL 为 −0.36 V。
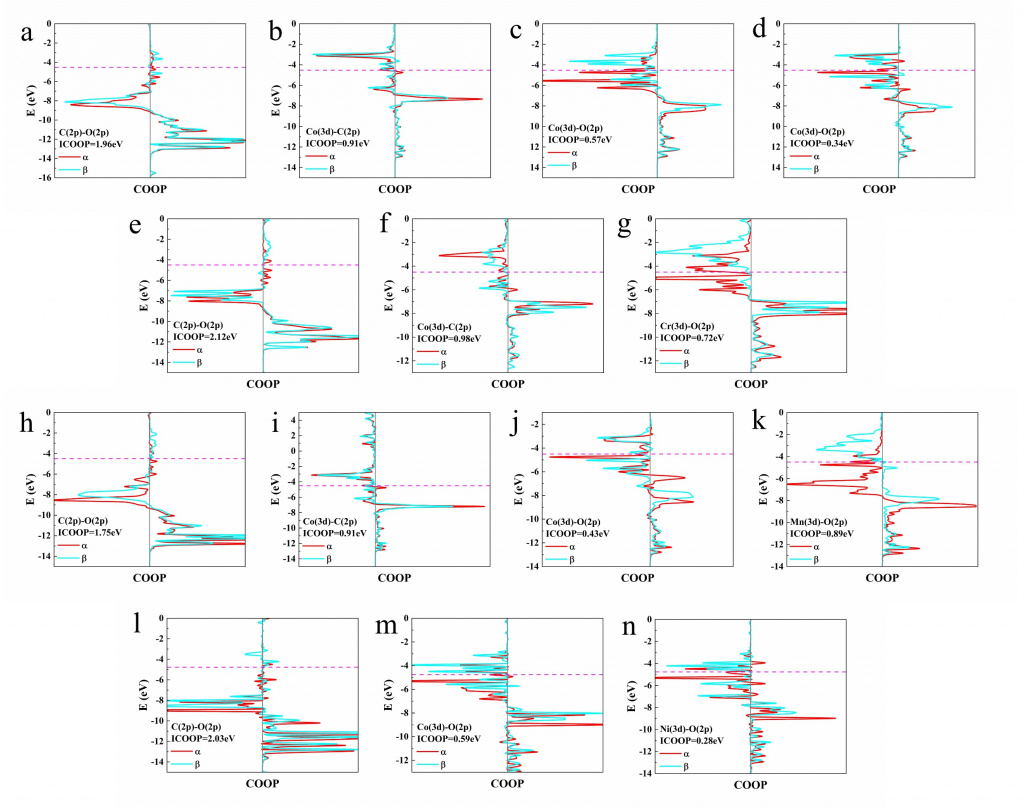
基于 Bader 电荷分析、PDOS 及 COOP计算,本研究阐明了 M1N3-M2N3@NGr-a 催化剂上的 CO2RR 机理:(1)两个 CO 分子在 DACs 上的强吸附作用有利于 C2 产物的形成;(2)该类 DACs 存在两种产物选择性调控机制,第一种机制主要源于催化剂对氧的强吸附能力,这可促使 *OC2H4 中间体中 C–O 键断裂,从而更有利于生成乙烯而非乙醇;第二种机制主要源于催化剂对乙烯的弱吸附作用,从而可提升乙烯的选择性。
总结
该研究从双原子位点构型、CO2 初始活化、关键中间体稳定到 C2 产物选择性调控,对 Co 基双原子氮掺杂石墨烯催化剂的 CO2RR 过程进行了较系统的理论分析。结果表明,a 构型整体优于 s 构型,在稳定性、CO2 活化能力和 C2 产物选择性方面均表现更好;其中,CoN3–NiN3@NGr-a 在乙醇生成路径上表现突出,CoN3–FeN3@NGr-a 在乙烯生成路径上更具优势。更重要的是,研究进一步揭示了不同 C2 产物形成背后的电子结构根源,为双原子催化剂的理性设计提供了有价值的理论参考。
从研究过程来看,AMS 不仅提供了二维周期体系建模和反应路径分析所需的计算支撑,也帮助实现了从结构稳定性评估到选择性机理解析的连贯研究流程。对于双原子催化剂这类结构复杂、路径分叉明显的体系而言,这样的计算平台能够有效提升研究效率,并为高性能 CO2RR 催化材料设计提供更加清晰的理论依据。
参考文献
- Theoretical study on Electrocatalytic Reduction of CO2 over Co based nitrogen doped graphene nanolayer supported diatomic catalyst, Physical Chemistry Chemical Physics, 2026, DOI:10.1039/D6CP00601A