
摘要
Hammett 方程通常用于描述取代基对金属有机骨架 (MOFs) 金属节点催化活性的远程电子效应。然而,该理论在 MOFs 催化剂上的应用经常遇到问题,因为它严重依赖于可移植性未知的经验参数。为了发展更普适的替代理论,国家纳米科学中心高兴发、孟幻,江西师范大学高雪皎等课题组,通过密度泛函理论计算,提出了配体轨道能量模型。该模型提供了一种简单的方法,近似描述远程电子取代基对 MOFs 金属节点催化活性的影响,并通过对文献中报道的结构-活性关系的广泛回顾,验证了对 MOFs 的普遍适用性。
在这种模型中,催化关键能垒与配体轨道能量呈近似线性关系,不依赖于经验参数,适用于具有不同配体和取代基的 MOFs 。与 Hammett 模型相比,通用性更高,允许研究者通过调节配体和取代基的电子性质来设计 MOFs 金属节点的催化活性。
配体轨道能量模型
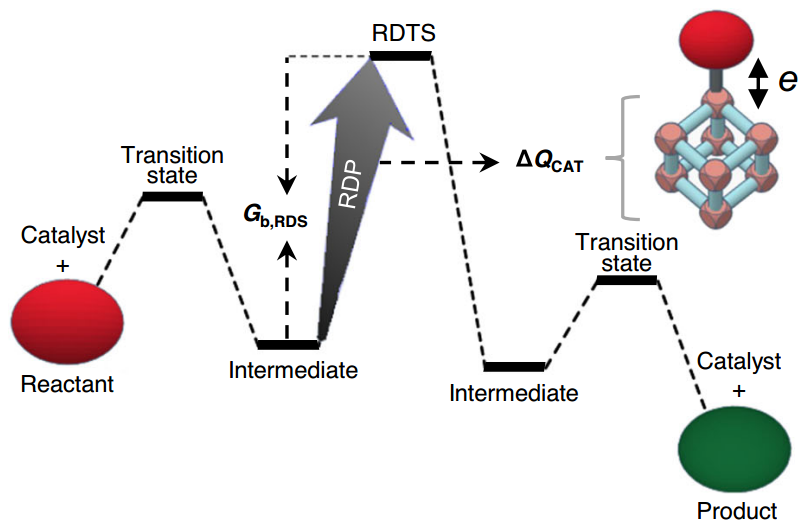
对 UiO-66-X (X=NH$_2$,H,O$_2$) 两种不同电子转移方向( MOFs 推/拉电子)的催化反应中的决速布进行了研究,发现电子取代基对“催化作用区域原子”的总电荷转移 ΔQ$_{CAT}$ 增强越多,则决速步自由能垒越低,从而 MOFs 金属节点催化活性也越大。
通过 AMS 软件 ADF 模块中的 ETS-NOCV 方法,分析催化剂基团与反应物之间的轨道相互作用,发现两类反应的主要轨道相互作用其实是相似的,即金属 d$_{z^2}$ 轨道和配体 p 轨道之间的相互作用。由于配体取代基与节点金属之间的远程 π-d 共轭,取代基通过调节金属的核外电子密度,即可调节金属的有效核荷(Z$_{eff}$),进而调节包括金属 d$_{z^2}$ 轨道在内的分子轨道的能量,实现对金属催化活性的调节。这种π-d共轭催化活性的取代基效应,有时被理解为远程共轭调节了节点金属的Lewis酸度。
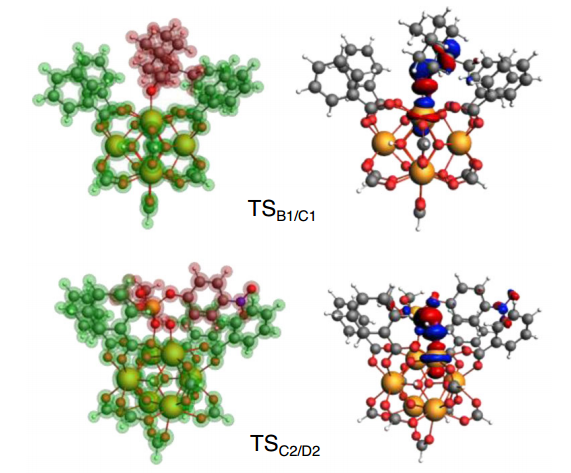
作者发现催化剂的前线轨道能量与配体的前线轨道能量具有良好的线性关系(因为UiO-66-X团簇的 HOMO 和 LUMO 都主要位于配体上)。通过数值线性拟合,建立了催化活性(以决速步自由能垒来表征)与催化剂前线轨道能量的关系。
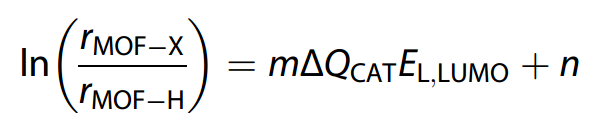
值得注意的是,尽管 ΔQ$_{CAT}$ 是理解模型背后机制的关键,但在使用配体轨道能量模型时,并不需要 ΔQ$_{CAT}$ 的确切值。同样配体前线轨道的精确能量也不是必需的,获得轨道能量的相对值就足以使用该模型。由于前线轨道能量是配体的固有性质,其相对值可以通过DFT计算或光电子能谱实验轻松可靠地获得。
该模型提供了一种简单的、通过配体取代基来调节MOFs的催化活性的方法。
参考文献
Remote substituent effects on catalytic activity of metalorganic frameworks: a linker orbital energy model, Zhenzhen Wang , Huan Meng , Xuejiao J. Gao, Jia-Jia Zheng and Xingfa Gao, npj Computational Materials (2023) 9:59